Advanced Therapy Medicinal Products: Clinical Development Challenges, Regulatory Evolution, and Strategic Considerations
Advanced Therapy Medicinal Products (ATMPs) are transforming modern medicine, but clinical development remains highly complex. This article explores key challenges in ATMP clinical trials, recent FDA and EMA regulatory updates, real-world success and failure cases, and how biotech leaders can select the right CRO for these advanced therapies.
Advanced Therapy Medicinal Products: Clinical Development Challenges, Regulatory Evolution, and Strategic Considerations
Advanced Therapy Medicinal Products: Clinical Development Challenges, Regulatory Evolution, and Strategic Considerations
Advanced Therapy Medicinal Products (ATMPs) – including gene therapies, cell therapies, and tissue-engineered products – represent one of the most profound shifts in modern medicine, with ATMP clinical trials playing a critical role in translating these innovations into approved therapies. Instead of targeting downstream symptoms, these therapies aim to correct or replace the biological mechanisms underlying disease.
Over the last decade, ATMPs have evolved from experimental academic concepts into a rapidly expanding therapeutic field. Both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have approved an increasing number of cell and gene therapies, and hundreds more are currently in clinical development.
Yet translating breakthrough biology into approved therapies remains exceptionally complex. Unlike traditional pharmaceuticals, ATMP development simultaneously requires mastery of clinical science, manufacturing, regulatory strategy, and translational biology.
For biotech leaders navigating this landscape, understanding both the opportunities and the risks is critical.
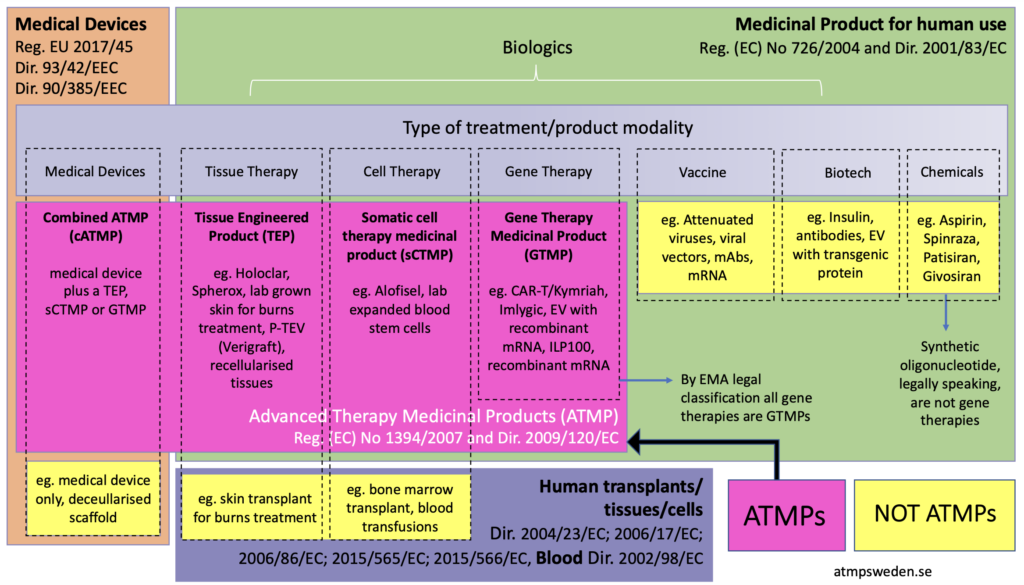
What qualifies as an ATMP?
Under European regulation (Regulation EC No 1394/2007), ATMPs are defined as therapies based on genes, cells, or engineered tissues. The category includes three main classes:
Gene therapy medicinal products – therapies delivering genetic material to modify or replace defective genes
Somatic cell therapy medicinal products – treatments using manipulated cells to restore or modify biological function
Tissue-engineered products – therapies designed to regenerate or repair damaged tissues
ATMP classification is well summarized by ATMP Sweden in the image below:
Each of these categories presents unique development challenges. Unlike small molecules, where the active substance is chemically defined, ATMPs often involve living biological systems, making characterization and consistency more difficult.
Recent ATMP successes and setbacks
The rapid expansion of ATMPs has produced several remarkable clinical successes — but also some notable failures. Together they illustrate both the promise and the complexity of this field.
Recent clinical successes
Several recently approved therapies demonstrate how transformative these technologies can be.
Examples include:
Kymriah (tisagenlecleucel) and Yescarta (axicabtagene ciloleucel), CAR-T cell therapies that have significantly improved outcomes for patients with refractory leukemia and lymphoma.
Luxturna, a gene therapy restoring functional vision in patients with inherited retinal dystrophy caused by RPE65 mutations.
Hemgenix, an AAV-based gene therapy for hemophilia B that demonstrated sustained factor IX expression after a single administration.
Casgevy, the first CRISPR-based gene editing therapy approved for sickle cell disease.
These therapies demonstrate that ATMPs can achieve outcomes that were previously impossible using conventional drugs.
Development setbacks
However, the ATMP field has also experienced significant failures.
Notable examples include:
Zynteglo initially struggled with regulatory delays and commercial uptake due to extremely high treatment costs.
Skysona raised safety concerns due to cases of secondary malignancies linked to lentiviral integration.
Several gene therapy programs in neuromuscular and metabolic diseases have failed in late-stage trials due to insufficient durability of gene expression or unexpected immune responses.
These experiences highlight a key lesson: scientific innovation alone does not guarantee clinical or commercial success.
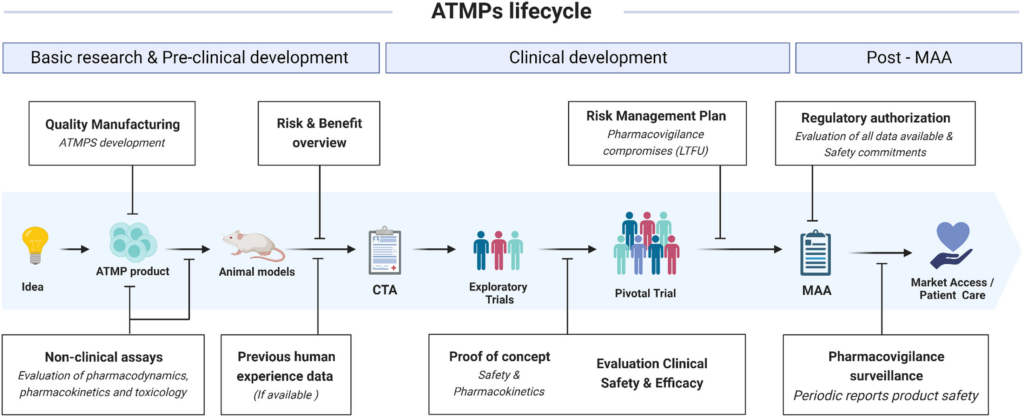
Key clinical development challenges in ATMP programs
Developing ATMPs presents a number of unique scientific and operational challenges. These include manufacturing and product consistency, design of ATMP clinical trials and long-term safety monitoring, as shown in the ATMPs lifecycle schematics below.
Manufacturing and product consistency
In ATMPs, the manufacturing process often defines the product itself. Small variations in cell expansion conditions, viral vector production, or purification methods can significantly influence therapeutic potency and safety.
Major challenges include:
maintaining batch-to-batch consistency
scaling manufacturing for commercial production
ensuring chain-of-identity and chain-of-custody for autologous therapies
establishing validated potency assays
These challenges make Chemistry, Manufacturing, and Controls (CMC) one of the most critical aspects of ATMP development.
Designing ATMP Clinical trials
ATMP clinical trials often target rare diseases or highly specialized patient populations, which limits traditional randomized study designs.
Developers frequently face challenges such as:
small patient populations
limited natural history data
ethical constraints around placebo arms
reliance on surrogate or biomarker endpoints
Both the FDA and EMA have acknowledged these challenges and allow adaptive trial designs, external control datasets, and natural history comparisons in appropriate situations.
Long-term safety monitoring
Gene therapies and cell therapies may produce long-lasting or permanent biological effects, which introduces additional safety considerations.
Regulators often require long-term follow-up programs to monitor for:
insertional mutagenesis
delayed immune reactions
secondary malignancies
persistence of engineered cells
The FDA recommends follow-up periods of up to 15 years for certain gene therapy products.
Regulatory evolution: FDA and EMA
Recognizing the transformative potential of ATMPs, regulatory agencies have developed dedicated frameworks to support innovation while maintaining patient safety.
In the United States, the U.S. Food and Drug Administration introduced the Regenerative Medicine Advanced Therapy (RMAT) designation, designed to accelerate development of therapies addressing serious diseases with preliminary evidence of efficacy.
RMAT designation provides:
early and frequent interaction with the FDA
eligibility for accelerated approval pathways
guidance on efficient clinical trial design
Similarly, the European Medicines Agency regulates advanced therapies through the Committee for Advanced Therapies (CAT) and supports development through the PRIME (PRIority MEdicines) scheme.
Recent regulatory discussions in both jurisdictions emphasize:
increased flexibility in CMC requirements for early development
adaptive trial designs for small patient populations
earlier engagement between regulators and developers
These developments reflect a growing recognition that traditional drug development frameworks do not always fit advanced therapies.
How to select a CRO for ATMP clinical trials
For biotech companies, selecting a CRO for ATMP clinical trials is not merely an operational choice – it is a strategic one. Several factors should be carefully evaluated.
Early-phase expertise
Many ATMP programs begin with first-in-human trials involving complex safety considerations. CRO partners should have experience managing:
dose-escalation studies
complex safety monitoring
adaptive trial designs
Translational science integration
ATMP clinical trials rely heavily on translational endpoints. Important capabilities include:
biomarker development
biodistribution studies
immune response monitoring
Operational logistics
Advanced therapies often require specialized clinical logistics, including cryogenic transport and coordination with manufacturing facilities.
Regulatory strategy support
Given the evolving regulatory landscape, CROs must also support interactions with regulatory agencies and help navigate accelerated pathways such as RMAT or PRIME.
Looking ahead
The next generation of ATMP innovation is already emerging.
Key technologies shaping the future include:
CRISPR and other gene-editing platforms
allogeneic “off-the-shelf” cell therapies
next-generation CAR-T and CAR-NK therapies
in vivo gene delivery technologies
As these platforms mature, ATMPs may expand beyond rare diseases into broader indications such as cardiovascular disease, autoimmune disorders, and neurodegenerative conditions.
For biotech and pharmaceutical leaders, the opportunity is enormous — but success requires more than breakthrough science. It demands integrated expertise in clinical development, manufacturing, regulatory strategy, and operational execution.
Advanced therapies are not simply another drug modality. They represent a fundamental shift in how medicine treats disease. And for organizations able to navigate this complexity, the potential impact on patients – and on the future of medicine – is extraordinary.
Planning ATMP clinical trials in Europe? Let’s chat! Contact our business development team and schedule a call to know how APICES CRO handles the acceleration of study timelines and generation of valuable data according to plan.
Get the latest clinical insights delivered to your feed or inbox!
Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I...
Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based...
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
")
")