EU MDR Clinical Investigations: Article 62 and Article 82 Pathways Explained (2026)
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
EU MDR Clinical Investigations: Article 62 and Article 82 Pathways Explained (2026)
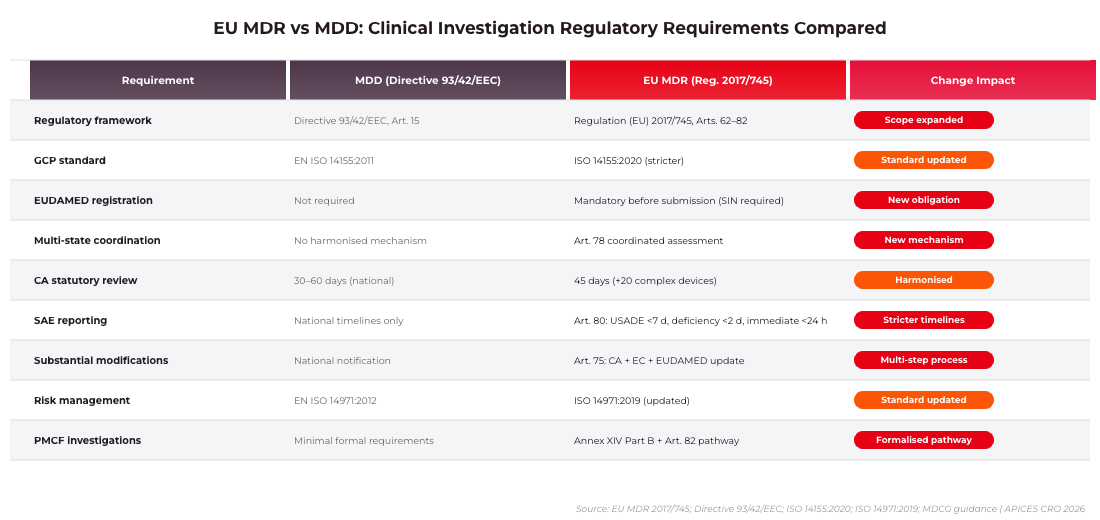
Key Takeaways: EU MDR 2017/745 establishes two distinct clinical investigation pathways for Medtech sponsors: Article 62 governs pre-market and new-indication investigations of investigational devices, while Article 82 covers PMCF and other post-market clinical investigations. Each pathway carries different competent authority timelines, Ethics Committee requirements, and EUDAMED obligations. Sponsors who misclassify their study type face rejection, delays of 6–18 months, and potential data integrity issues. In 2026, all clinical investigations must be registered in EUDAMED, and coordinated assessment under Article 78 is now fully operational.
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
What Qualifies as a Clinical Investigation Under EU MDR?
Article 2(45) of EU MDR defines a clinical investigation as “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device.” The scope is broader than under MDD. Any study that prospectively collects clinical data on an investigational device, or on a CE-marked device used outside its intended purpose, falls within the Chapter VI framework.
Not all device studies are clinical investigations. Post-Market Clinical Follow-Up studies conducted through literature review, device registries, or patient questionnaires are PMCF activities governed by Annex XIV Part B and MDR Article 83 – not clinical investigations. The distinction matters: Annex XIV Part B studies do not require competent authority submission or EUDAMED clinical investigation registration, whereas Article 62 and Article 82 investigations do.
Devices Covered by Chapter VI
Investigational devices – devices not yet CE-marked, or CE-marked devices being evaluated for a new intended purpose or indication.
CE-marked devices in PMCF investigations – CE-marked devices studied under a formal clinical investigation protocol for PMCF purposes (rather than through registries or chart review).
Devices in combination with an IMP – where the device component is investigational, the device-specific requirements of Chapter VI apply alongside applicable pharmaceutical legislation.
EU MDR Article 62 Submission Process: 7-step pathway from CIP development to first subject. Source: EU MDR 2017/745 Arts. 62–80; ISO 14155:2020 | APICES CRO 2026
The Article 62 Pathway: Pre-Market and New Indication Investigations
Article 62 of EU MDR applies to clinical investigations conducted to demonstrate conformity with the general safety and performance requirements of Annex I. In practice, this covers three scenarios: first-in-human (FIH) studies, pivotal investigations supporting a CE conformity assessment, and studies of CE-marked devices for intended purposes not yet covered by the existing Certificate of Conformity.
The Article 62 procedure requires sponsors to submit an application via EUDAMED (Regulation (EU) 2017/745, Article 70(1)) and obtain both Ethics Committee (EC) approval and competent authority (CA) authorisation before study initiation. Under the coordinated assessment mechanism introduced by Article 78, sponsors may designate one member state as the Coordinating Member State (CMS) to lead the CA review, with Concerned Member States conducting parallel national assessments. This mechanism has been mandatory for multi-state investigations since its operational launch.
Article 62 Application Requirements
Clinical Investigation Plan (CIP) – Must conform to the requirements of Annex XV, Chapter II, including design rationale, risk-benefit justification, statistical methodology, and endpoint hierarchy.
Investigator’s Brochure (IB) or Device Description – For investigational devices, a full technical description including materials, manufacturing methods, and non-clinical test results.
Risk management documentation – Risk management file summary per ISO 14971:2019.
Insurance or indemnification documentation – Proof of sponsor liability coverage per Article 69(1) of EU MDR.
EUDAMED registration – All clinical investigations must be assigned a Single Identification Number (SIN) via EUDAMED before submission to member states. The SIN is a prerequisite for any CA or EC submission as of 2025.
GCP compliance declaration – Sponsor declaration of compliance with ISO 14155:2020 (Good Clinical Practice for clinical investigations of medical devices).
Competent Authority Timelines Under Article 65
Article 65 sets the statutory assessment timeline at 45 days from receipt of a valid application (the “Day 0” date). Member states may extend this timeline by 20 additional days for devices incorporating an ancillary medicinal substance or materials of animal or human origin, for a maximum of 65 days. In practice, CA timelines differ substantially across member states based on national implementation and regulatory authority capacity. Sponsors should build country-specific timelines into their study feasibility assessment from the earliest protocol development phase.
EU MDR vs MDD: Clinical Investigation Regulatory Requirements Compared. Source: Regulation (EU) 2017/745; Directive 93/42/EEC; ISO 14155:2020 | APICES CRO 2026
The Article 82 Pathway: PMCF and Other Post-Market Investigations
Article 82 of EU MDR covers clinical investigations that do not fall under Article 62 – principally, formal clinical investigations conducted for PMCF purposes on devices already CE-marked and used within their existing intended purpose. The Article 82 pathway was designed for scenarios where a sponsor needs to collect prospective, protocol-driven clinical evidence to fulfil PMCF obligations under Annex XIV Part B, but the study design requires participant informed consent, protocol-defined follow-up visits, or data collection beyond routine clinical care.
Under Article 82(1), member states may apply national procedural requirements to these investigations, provided such requirements do not impose an undue burden or alter the protections afforded to participants. This means that Article 82 studies are subject to national legislation in each member state where they are conducted. In Germany, Article 82 investigations fall under the Medizinprodukterecht-Durchführungsgesetz (MPDG). In Spain, they are regulated by Real Decreto 1090/2015 as adapted to MDR. In France, the Agence nationale de sécurité du médicament et des produits de santé (ANSM) processes Article 82 notifications through its dispositifs médicaux division.
Key Differences Between Article 62 and Article 82
Device status – Article 62 applies to investigational or new-indication devices; Article 82 applies to CE-marked devices used within their intended purpose for PMCF.
Competent authority role – Article 62 requires CA authorisation before study start; Article 82 requirements are set by national law and may allow notification-only or abbreviated review in some states.
Coordinated assessment – Article 78 coordinated assessment is mandatory for multi-state Article 62 investigations; it does not apply to Article 82 investigations.
EUDAMED registration – Both pathways require EUDAMED registration as of 2025, but the Article 82 registration is classified differently in the system.
Serious adverse event reporting – Article 62 investigations require SAE/SADE reporting to competent authorities under Articles 69 and 80. Article 82 investigations are subject to national SAE reporting requirements, which may differ from the Article 69 timelines (24 hours for device deficiency posing risk, 7 days for serious incident).
Article 74: Clinical Investigations in Emergency Situations
Article 74 provides a derogation from the standard informed consent requirements in Articles 63(2) and 63(3) for clinical investigations conducted in life-threatening or emergency situations. Where it is clinically impossible to obtain prior informed consent, Article 74 permits the investigation to proceed provided the Ethics Committee and competent authority have authorised this approach in advance, and provided consent is sought from the subject or their legally authorised representative at the earliest possible opportunity.
This provision is most relevant for investigations of devices used in cardiac arrest, stroke, major trauma, or other emergent conditions. Sponsors planning emergency-setting investigations should build the Article 74 justification into the Ethics Committee submission from the outset, as post-hoc justification is not acceptable under the regulation.
EUDAMED Registration Requirements in 2026
EUDAMED’s clinical investigation module has been fully operational for new submissions since mid-2025 following the release of EUDAMED version 4.0. All clinical investigations under both Article 62 and Article 82 must be registered before submission to any member state competent authority. The Single Identification Number (SIN) generated by EUDAMED must appear on all national submissions, Ethics Committee applications, informed consent forms, and CIP headers.
The European Commission’s Medical Device Coordination Group (MDCG) published MDCG 2021-8 Rev. 1 in 2024 to clarify EUDAMED registration obligations for clinical investigations, addressing transition provisions for investigations that began under MDD and transitioned to MDR status. Sponsors with ongoing legacy investigations should verify their EUDAMED registration status against the MDCG 2021-8 Rev. 1 checklist.
Substantial Modifications Under Article 75
Article 75 requires sponsors to notify the relevant member state competent authority and Ethics Committee of any substantial modification to a clinical investigation before implementing the change. A modification is substantial if it is likely to have a significant impact on the safety or rights of the subjects, or on the reliability or robustness of the clinical data generated. Sponsors must update the EUDAMED registration concurrent with or prior to the national notification. Common substantial modifications include: protocol amendments affecting primary endpoints, changes to inclusion/exclusion criteria affecting subject risk profile, device modifications affecting intended purpose or risk classification, and changes to the principal investigator at any site.
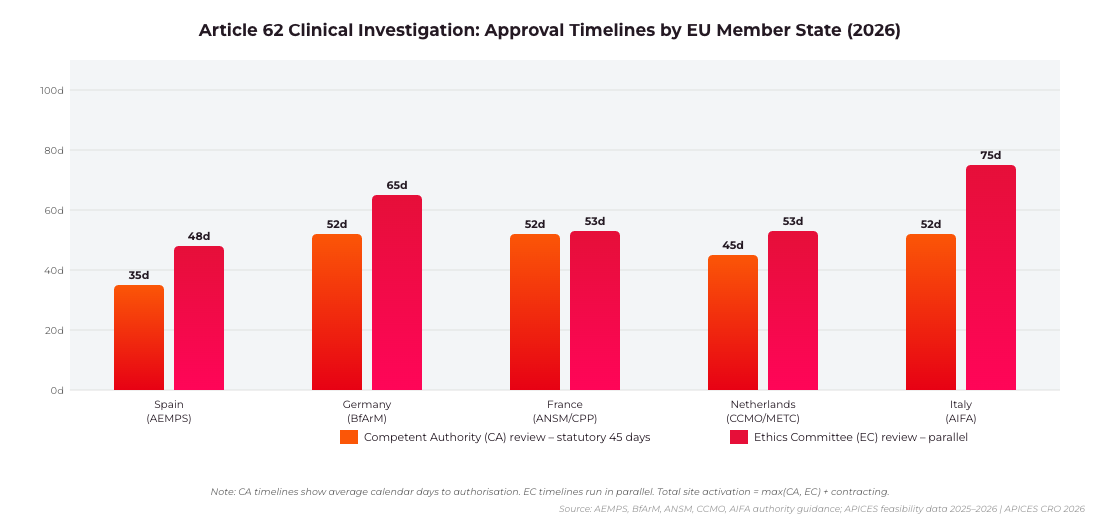
Article 62 Approval Timelines by EU Member State: CA review vs Ethics Committee review in days. Source: AEMPS, BfArM, ANSM, CCMO, AIFA guidance; APICES feasibility data 2025–2026 | APICES CRO 2026
Country-Specific Considerations for Multi-State Investigations
Spain remains one of the fastest competent authority jurisdictions in the EU for Article 62 clinical investigations. The Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) typically issues CA authorisation within 30–40 days of a valid application, making Spain a preferred first-in-class submission country in European multi-site investigations. For Article 82 PMCF investigations, Spain accepts a simplified notification with a 30-day acknowledgement window.
Germany processes Article 62 applications through the Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), with standard CA review running 45–60 days. German Ethics Committees operate under the consolidated Ethikkommissionen framework and typically issue opinions within 60 days of a complete submission.
France processes Article 62 applications through the ANSM with Comité de Protection des Personnes (CPP) approval required as a prerequisite. Since the implementation of Loi Jardé (Law No. 2012-300) as updated for MDR alignment, interventional investigations require CPP approval within a 45-day review period before ANSM authorisation. The Netherlands (CCMO – Central Committee on Research Involving Human Subjects) requires combined METC and CCMO review for most Article 62 investigations, with a statutory 40-day review period. In practice, total approval timelines including site contracting in Spain average 3–5 months for Article 62 studies and 2–3 months for Article 82 studies.
Frequently Asked Questions
What is the difference between Article 62 and Article 82 in EU MDR?
Article 62 of EU MDR 2017/745 governs clinical investigations of investigational devices – devices that are not yet CE-marked, or CE-marked devices being evaluated for a new intended purpose. Article 82 governs other clinical investigations, principally PMCF investigations of CE-marked devices used within their existing intended purpose. Article 62 requires competent authority authorisation and mandatory Ethics Committee approval before study start, following the procedures in Articles 62–80. Article 82 investigations follow national procedural requirements, which vary by member state, and do not require the coordinated assessment mechanism of Article 78.
Does a PMCF study always require a formal clinical investigation?
No. PMCF activities under Annex XIV Part B of EU MDR include both formal clinical investigations (which require Ethics Committee approval, EUDAMED registration, and CA notification or authorisation) and non-interventional activities such as literature reviews, device registries, post-market surveillance surveys, and retrospective chart reviews. Only protocol-driven prospective studies that involve participant informed consent, additional procedures beyond standard care, or collection of data for a pre-defined research purpose constitute a formal clinical investigation. Sponsors should document their PMCF classification decision in the PMCF Plan, referencing the criteria in MDCG 2020-7 Rev. 1 (PMCF Studies).
When is the EUDAMED Single Identification Number (SIN) required?
As of mid-2025, EUDAMED SIN registration is required for all clinical investigations under both Article 62 and Article 82 before any national submission is made to a competent authority or Ethics Committee. The SIN must appear on all application documents. The EUDAMED clinical investigation module issues a SIN immediately upon registration of basic study information. Sponsors should allow 2–3 business days for EUDAMED account provisioning if registering for the first time, as actor registration is a prerequisite for clinical investigation module access.
What are the serious adverse event reporting timelines for EU MDR clinical investigations?
Under Article 80 of EU MDR, sponsors must report serious adverse device effects (SADE) and unanticipated serious adverse device effects (USADE) without delay; device deficiencies that could have led to a SADE must be reported within 2 days of identification. Full USADE reports must follow within 7 days of the initial report. Device deficiencies posing immediate risk must be reported within 24 hours. All reports are submitted through EUDAMED’s clinical investigation module. These timelines are stricter than MDD-era requirements and require sponsors to establish robust study-specific safety monitoring and reporting SOPs before site activation.
How does the Article 78 coordinated assessment work in practice?
Article 78 of EU MDR establishes a coordinated assessment procedure for multi-state clinical investigations. The sponsor designates one member state as the Coordinating Member State (CMS), which leads on reviewing the clinical and scientific aspects of the application. Concerned Member States conduct parallel national assessments. The CMS issues a single assessment report within the statutory timeline (45 days, extendable to 65 days for complex devices). Each Concerned Member State then issues a national decision based on the coordinated report and its own review of national-specific elements. Coordinated assessment reduces duplication of scientific review across states but does not eliminate national Ethics Committee approval timelines or site contracting requirements.
Planning Your EU MDR Clinical Investigation: Next Steps
Whether your programme requires an Article 62 pivotal investigation, an Article 82 PMCF protocol, or a non-interventional PMCF study under Annex XIV Part B, the regulatory pathway decisions made early in protocol development determine timelines, budget, and data quality for the entire investigation. At APICES CRO, our medical devices team provides regulatory strategy, CIP writing, EUDAMED submissions, Ethics Committee navigation, and full clinical operations support across Spain, France, Germany, and the Netherlands. If you are planning a clinical investigation under EU MDR 2017/745, contact our regulatory team to schedule a no-obligation feasibility assessment.
Get the latest clinical insights delivered to your feed or inbox!
Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I...
Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based...
Complete guide to Phase I oncology trials in Western Europe in 2026: CTIS submissions, country selection (Spain, France, Germany, Netherlands), site criteria, study design, and CRO selection for biotech sponsors.
")
")