Phase I Oncology Trials in Western Europe: The Complete Execution Guide for Biotech Sponsors (2026 Edition)
Complete guide to Phase I oncology trials in Western Europe in 2026: CTIS submissions, country selection (Spain, France, Germany, Netherlands), site criteria, study design, and CRO selection for biotech sponsors.
Phase I Oncology Trials in Western Europe: The Complete Execution Guide for Biotech Sponsors (2026 Edition)
Key Takeaways: Western Europe remains the most credible environment for Phase I oncology trials in 2026. CTIS has standardised submissions but country-level execution still determines start-up speed. Spain, France, the Netherlands, and Germany each offer distinct advantages depending on your indication, site requirements, and timeline. CRO selection should be based on lived execution experience, not feasibility spreadsheets.
If you are a biotech CEO or CMO preparing your first Phase I oncology program in Europe, the decisions you make in the next 90 days – which countries, which sites, which CRO – will determine whether your study initiates in 12 months or 20. The EU’s Clinical Trials Regulation (Regulation EU No 536/2014, the “EU CTR”) and its associated Clinical Trials Information System (CTIS) have standardised the submission architecture across Europe. But standardised submissions do not mean standardised execution. Country-by-country differences in site infrastructure, patient access, regulatory culture, and institutional contracting still drive the variance between a well-planned Phase I program and an expensive correction.
This guide covers the complete execution landscape for Phase I oncology trials in Western Europe in 2026: the regulatory framework under CTIS, country selection logic, site considerations specific to oncology Phase I, study design practicalities, and CRO selection criteria. It is written from the perspective of what actually happens once a protocol is approved – not what looks optimal in a feasibility model.
The EU CTR and CTIS: What Has Actually Changed for Phase I Sponsors
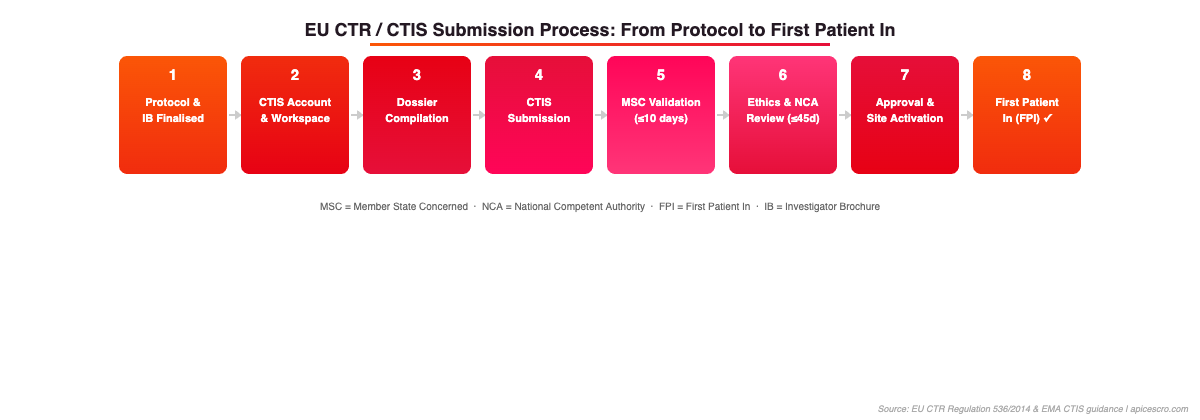
The EU CTR became mandatory for all new clinical trial applications on 31 January 2025, completing a three-year transition. Every Phase I oncology trial launching in Europe now requires submission via CTIS – the centralised EU portal managed by the European Medicines Agency (EMA). Understanding what CTIS changes operationally matters more than understanding its architecture.
CTIS introduces a two-part submission structure. Part I covers the scientific and regulatory dossier – the Investigational Medicinal Product Dossier (IMPD), the protocol, and the risk-benefit assessment. It is assessed jointly by the Reference Member State and Concerned Member States. Part II covers country-specific components: ethics committee approvals, patient-facing documents (consent forms, patient information sheets), and local regulatory submissions. Part II is assessed separately by each Member State.
For Phase I sponsors, the practical implications are significant. The Part I assessment window is 30 days (extendable to 45 for complex trials including Phase I oncology). Part II assessment runs in parallel and must be completed within 30 days of Part I approval in each country. In practice, countries with well-resourced ethics committees – notably the Netherlands, Spain, and France – consistently meet or outperform these windows. Countries with more fragmented ethics infrastructure can add 4–8 additional weeks to your actual study start date even after regulatory approval.
A critical change under the EU CTR is the public disclosure requirement. CTIS publishes trial information – including the protocol – on the public portal once a study reaches the start of the Part I assessment. For early-phase oncology programs where mechanism-of-action confidentiality matters, this requires careful timing of CTIS submissions relative to patent filings and competitive intelligence considerations. Sponsors should align legal, IP, and regulatory teams before CTIS submission, not after.
Figure 1: EU CTR/CTIS Submission Process – 8 steps from protocol finalisation to First Patient In. Source: EU CTR Regulation 536/2014 & EMA CTIS guidance.
Country Selection for Phase I Oncology: Beyond the Spreadsheet
Country selection for Phase I oncology is rarely a pure regulatory efficiency question. It is a function of five interlocking variables: regulatory submission speed, ethics committee quality and predictability, Phase I unit infrastructure, oncology patient access and molecular pre-screening capability, and contracting and payment norms.
Spain
Spain is consistently one of the fastest Phase I environments in Western Europe. The Spanish Agency of Medicines and Medical Devices (AEMPS) processes CTIS Part I submissions efficiently, and the Spanish ethics committee reform has reduced the variability that characterised pre-CTR approvals. Spanish Phase I oncology units – particularly at Hospital Universitario Vall d’Hebron (Barcelona), Hospital Universitario 12 de Octubre (Madrid), Hospital Universitario HM CIOCC, and Instituto Oncológico Baselga (IOB) – have strong track records in first-in-human and dose-escalation studies. Spain also offers competitive site costs relative to Northern Europe and above-average PI engagement for early-phase programs. The main risk is PI availability: the best Phase I PIs are heavily competed for, and feasibility conversations should happen at PI level very early in the planning process.
France
France’s Phase I landscape has strengthened since the integration of CTIS and the rationalisation of the ethics committee structure under the Comité de Protection des Personnes (CPP) system. Key Phase I oncology units include Institut Gustave Roussy (IGR, Villejuif), Institut Curie, Centre Léon Bérard (Lyon), and CLCC sites with dedicated early-phase departments. France has particular strengths in immuno-oncology, ADCs, and haematological malignancies. The HAS-regulated access to tumour molecular profiling programmes (SHIVA, AcSé) gives French sites robust biomarker-stratified pre-screening infrastructure. Contracting at French academic institutions can be slow (90–120 days is not unusual), which should be factored into start-up timelines from the outset.
The Netherlands
The Netherlands consistently records some of the fastest CTIS Part II completion times in the EU. The Central Committee on Research Involving Human Subjects (CCMO) runs an efficient centralised review process with a strong track record of meeting statutory timelines. The Netherlands Cancer Institute (NKI-AVL, Amsterdam), Erasmus MC Cancer Institute (Rotterdam), and Radboud UMC (Nijmegen) offer Phase I units with high scientific engagement and strong molecular pathology infrastructure. The Netherlands is particularly well-suited for first-in-human studies where early PK/PD characterisation is critical: Dutch clinical pharmacology capabilities in academic centres are among the strongest in Europe. Site costs are higher than Southern Europe, but contracting timelines tend to be shorter than France or Italy.
Germany
Germany offers the largest patient pool in Western Europe for oncology studies, but Phase I execution has historically been complicated by the dual-authority structure (BfArM for most products, Paul-Ehrlich-Institut for biologics and advanced therapies) and by a decentralised ethics committee landscape. Institutions with well-established Phase I units – Universitätsklinikum Heidelberg, Universitätsklinikum Köln, Charité (Berlin), and TU München – can perform very well for complex early-phase studies. For sponsors prioritising speed in the FiH single-ascending-dose phase, Germany may not be the optimal first country. A common strategy is to initiate FiH in Spain or the Netherlands and add Germany for the expansion cohort or Phase IIa portion of a seamless design.
Belgium and Switzerland
Belgium (FAMHP/AFMPS as competent authority; central ethics committee for Phase I) and Switzerland (Swissmedic, outside CTIS as a non-EU country) round out the Western European Phase I landscape. Belgium’s Hôpital Universitaire de Bruxelles (HUB, formerly Institut Jules Bordet) and UZ Leuven have strong Phase I track records in solid tumours and haematology respectively. Switzerland offers fast Swissmedic review (typically 30 days for Phase I) and exceptional site quality at CHUV (Lausanne), USZ (Zürich), and Inselspital (Bern). Including Switzerland adds a separate filing but can meaningfully accelerate overall programme timelines when EU regulatory processes encounter delays.
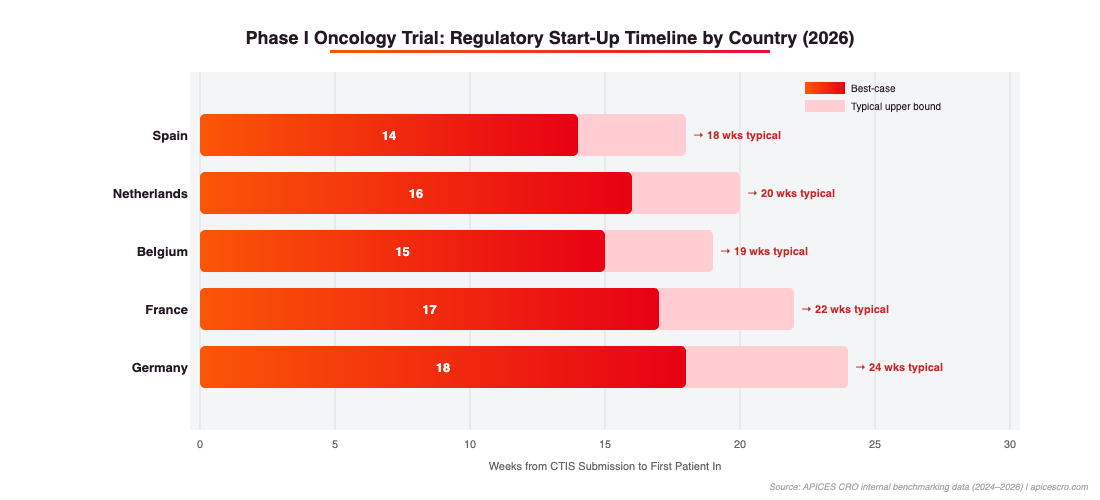
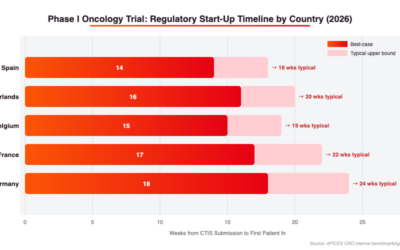
Figure 2: Regulatory start-up timelines by country (weeks from CTIS submission to First Patient In). Spain consistently delivers the fastest FPI. Source: APICES CRO internal benchmarking data 2024–2026.
Phase I Site Selection: What Actually Predicts Performance
Feasibility surveys are a necessary but insufficient basis for site selection in Phase I oncology. The question is not whether a site says they can recruit – it is whether the infrastructure behind that answer is real.
For Phase I oncology, the most predictive site performance indicators are:
Dedicated Phase I unit infrastructure: Dedicated inpatient beds or day-unit capacity for DLT observation windows, 24-hour nursing coverage for safety monitoring, and on-site pharmacy with experienced IMP reconstitution capability.
Molecular tumour board and biomarker pre-screening infrastructure: For biomarker-selected studies – which is the majority of modern Phase I oncology – the site’s ability to process FFPE samples, run NGS panels, and provide pre-screened patient lists before study initiation is a major differentiator. Sites without this capability create 6–12 week delays at the patient identification stage.
PI availability and competing trial load: The best Phase I PIs in Europe are typically running 8–15 active early-phase studies simultaneously. Assess PI availability directly – not through institutional business development teams.
Historical activation speed on comparable studies: Ask for the site’s actual activation timelines on their last three Phase I oncology studies – time from site selection to first patient enrolled. This is a direct performance indicator that feasibility surveys do not capture.
Contracting and finance office track record: Site contracting is frequently the longest component of the start-up critical path. Sites with dedicated clinical trials offices can activate in 6–8 weeks after ethics approval; sites without them can take 16–20 weeks.
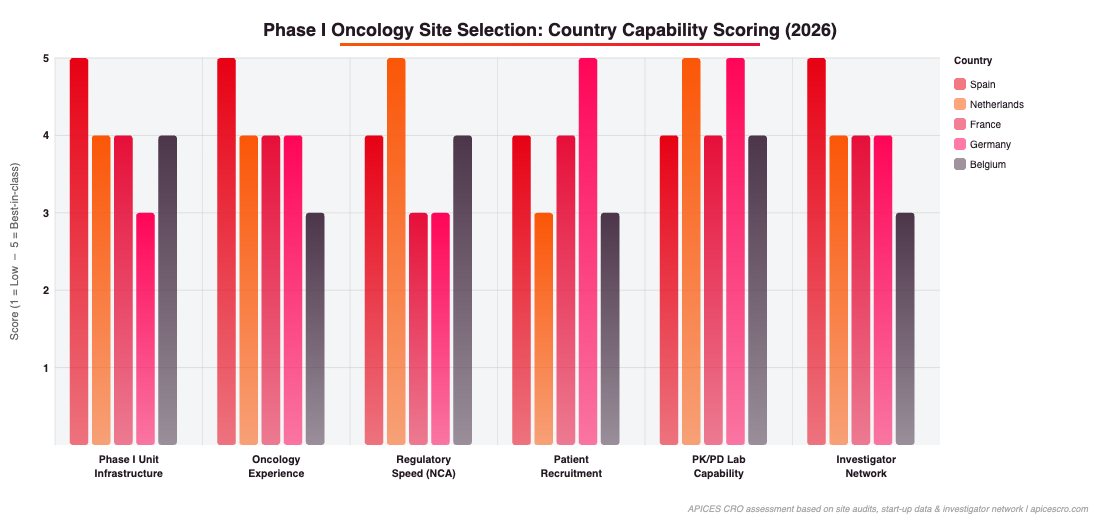
Figure 3: Country capability scoring across six Phase I site selection criteria. Spain leads on Phase I unit infrastructure and oncology experience; Netherlands on regulatory speed and PK/PD capability. Source: APICES CRO site audits and investigator network data.
Study Design Considerations for the EU Phase I Context
Phase I oncology study design choices have operational consequences in the European regulatory environment that are not always visible at the protocol design stage.
The choice of dose escalation design – 3+3, BOIN (Bayesian Optimal INterval), mTPI-2, or ROMI – affects both the number of dose levels and the number of patients per cohort, which directly impacts the number of sites needed and the complexity of the DSMB/IDMC charter. EMA’s scientific guidelines increasingly support model-based designs (the ICH E20 guideline reached Step 2b in June 2025), and leading European Phase I sites are familiar with BOIN and BLRM designs.
Seamless Phase I/II designs require careful pre-planning under the EU CTR. Protocol amendments under CTIS are subject to substantial modification procedures which can take 30–90 days. If your Phase I expansion cohort criteria or RP2D decision rules may evolve, these should be pre-specified in the original protocol as decision-tree options rather than addressed through amendments mid-study.
PK/PD sampling schedules must be aligned with the practical capabilities of each site’s clinical pharmacology team. Sparse sampling designs using population PK modelling are increasingly acceptable to EMA for early Phase I programmes and significantly reduce the operational burden on sites.
CRO Selection for Phase I Oncology in Europe
The CRO selection process for Phase I oncology in Europe is frequently over-indexed on price and under-indexed on execution capability. For a capital-constrained biotech, the cost of a CRO switch mid-study – in burned runway, regulatory delays, and lost investor confidence – is substantially higher than the cost difference between CRO bids.
The attributes that distinguish genuinely capable Phase I CROs from those that present well in RFP responses are:
Direct PI relationships at the sites you are targeting: Not managed relationships through a site network contract – direct, active relationships with the PIs who will run your study. Ask for PI references you can speak to directly.
Senior oversight that stays with the study: In Phase I oncology, the decisions that affect feasibility and timeline are made in the first 90 days. If the CRO team that wins your study hands off to a junior project management layer after kick-off, you lose the experience precisely when you need it most.
CTIS operational experience – not just familiarity: Ask specifically how many CTIS submissions the CRO has completed and what their average time from CTIS submission to first patient enrolled has been in the last 12 months for Phase I oncology studies.
Realistic feasibility: A CRO that challenges your country and site assumptions before contracts are signed is protecting your program. A CRO that validates your assumptions to win the contract is creating risk you will pay for later.
Frequently Asked Questions
How long does a Phase I oncology CTIS submission take from first submission to first patient enrolled in Western Europe?
For a 2–3 country Phase I oncology study in Western Europe, a realistic timeline from CTIS submission to first patient enrolled is 7–11 months. The CTIS assessment (Part I + Part II) typically takes 2–3 months when well-prepared. Site activation – contracting, ethics local sign-off, staff training, IMP availability – accounts for the remaining 4–8 months and is where most delays occur.
Can I run a Phase I first-in-human study in a single EU country?
Single-country FiH studies are common and operationally simpler. Spain and the Netherlands are the most frequently chosen single-country FiH environments in Western Europe because of their combination of regulatory speed, Phase I unit quality, and PK/PD infrastructure. Additional countries are typically added at the expansion cohort or seamless Phase IIa stage.
Does the EU CTR require a DSMB for Phase I oncology studies?
The EU CTR does not mandate a DSMB for all Phase I studies, but EMA guidance and ICH E6(R3) expect a risk-proportionate approach to independent safety oversight. For FiH studies, an independent DSMB with a pre-defined charter specifying dose escalation decisions, stopping rules, and DLT review procedures is considered best practice and will be scrutinised by ethics committees. The DSMB charter should be finalised before CTIS submission.
How many sites are needed for a Phase I oncology dose-escalation study in Western Europe?
For a standard FiH SAD/MAD Phase I using BOIN or 3+3 design with 4–6 dose levels and a small expansion cohort (15–30 patients), 3–5 sites across 2–3 countries is typically sufficient. More sites do not accelerate Phase I dose-escalation – they add coordination complexity. Once the study moves into an expansion cohort or seamless Phase IIa, 6–10 sites may be appropriate.
How does CTIS affect protocol confidentiality for a Phase I programme?
Under EU CTR Article 81, certain trial information becomes publicly available via the CTIS public portal. The protocol synopsis, IMP details, and primary endpoints are disclosed. Sponsors can request deferral of specific commercially sensitive information under EU CTR Article 81(4), but deferrals are limited in scope and duration. IP counsel should be involved in CTIS submission planning, and patent applications should be filed before CTIS submission wherever possible.
Running a Phase I oncology study in Western Europe requires the kind of execution experience that only comes from having done it. APICES supports biotech and pharma sponsors through every stage of early clinical development in Europe: from country and site selection to CTIS submission, Phase I unit activation, and first patient enrolled. If you are preparing a Phase I programme or want a realistic second opinion on how your current plan is set up, explore our Early Clinical Development offering or contact our team directly.
Get the latest clinical insights delivered to your feed or inbox!
Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I...
Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based...
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
")
")