Phase I Dose Escalation Design in the EU: ICH E8, EMA Guidelines and FDA Alignment for First-in-Human Studies
Phase I Dose Escalation Design in the EU: ICH E8, EMA Guidelines and FDA Alignment for First-in-Human Studies Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based […]
Phase I Dose Escalation Design in the EU: ICH E8, EMA Guidelines and FDA Alignment for First-in-Human Studies
Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based approaches such as BOIN and mTPI-2, which are now acceptable to EMA and reduce the risk of exposing patients to sub-therapeutic or toxic doses. Starting dose selection via MABEL (Minimum Anticipated Biological Effect Level) is mandatory for biologics and high-risk small molecules. The EU Clinical Trial Application for Phase I studies must include a detailed dose-escalation plan, DSMB/DMC charter, and stopping rules that comply with ICH E6(R3) GCP requirements.
Phase I dose escalation design in the EU has undergone significant evolution over the past five years. ICH E8(R1), finalised in 2021 and reflected in EMA guidance updates through 2024–2025, repositioned early clinical trials as hypothesis-testing experiments requiring fit-for-purpose statistical frameworks – not merely safety screening exercises. For sponsors planning first-in-human (FIH) studies in the European Union, understanding the regulatory architecture that governs phase I dose escalation design is no longer optional: CTIS submission requirements, EMA Scientific Advice timelines, and DSMB charter expectations all depend on it. This guide sets out the current EU regulatory framework, compares the principal dose escalation designs, and addresses the practical steps needed for a compliant EU Clinical Trial Application.
Regulatory Framework for Phase I Dose Escalation Design in the EU
ICH E8(R1) and EMA Alignment
ICH E8(R1) – adopted by EMA’s CHMP in November 2021 – introduced the concept of Quality by Design (QbD) for clinical studies. For Phase I dose escalation, this means that the choice of design (statistical or rule-based), the definition of dose-limiting toxicity (DLT), the observation window, and the stopping rules must all be pre-specified and justified in the protocol as integral elements of the study’s Quality Management Plan.
EMA’s existing guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials (EMA/CHMP/SWP/28367/07 Rev.1, 2017) remains the primary reference for FIH-specific requirements. It applies to all IMPs entering FIH trials in the EU and addresses MABEL calculation, starting dose justification, and sentinel dosing. A revised version incorporating experience with cell and gene therapies is expected in 2025–2026.
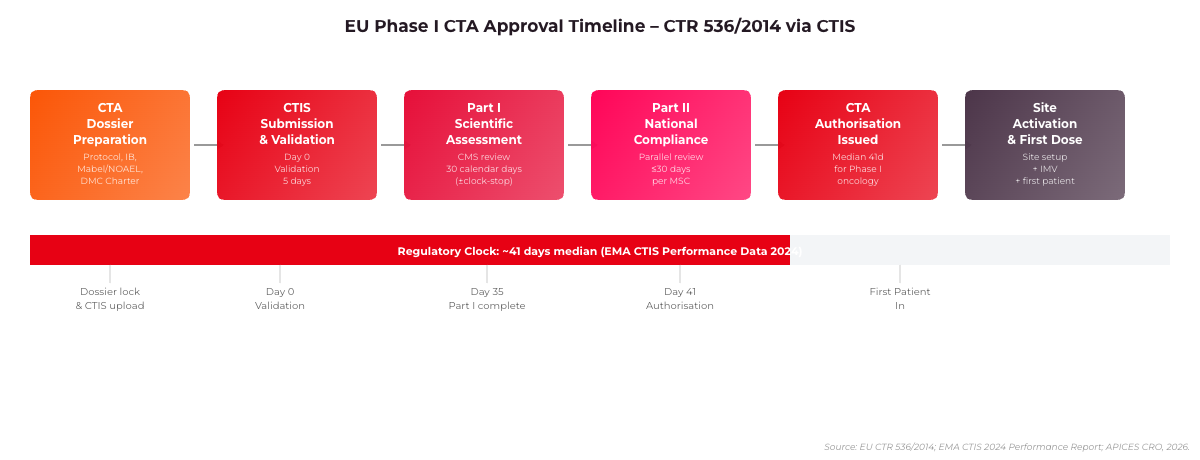
Figure 1: EU Phase I CTA Approval Timeline under CTR 536/2014. Median authorisation time is 41 days for multi-country Phase I oncology studies (EMA CTIS Performance Report, 2024).
EU Clinical Trial Regulation 536/2014 and CTIS Requirements
Since January 2023, all new clinical trial applications in the EU must be submitted via CTIS under Regulation 536/2014. For Phase I dose escalation studies, the CTIS submission package must include the following elements within the Part I Clinical Trial Authorisation dossier:
Protocol with full dose-escalation schema: The escalation algorithm (rule-based or model-based), DLT criteria with grading scale (CTCAE v5.0 or equivalent), and the DLT observation window must be explicitly stated. Regulatory authorities in the Coordinating Member State (CMS) will scrutinise these during the 30-day assessment window.
Investigator’s Brochure with MABEL/NOAEL justification: The starting dose section of the IB must cross-reference non-clinical pharmacology data and provide the MABEL derivation for biological IMPs. For small molecules, NOAEL-based derivations must include species-specific safety factors as per ICH S9 (oncology) or ICH S6 (biologics).
DSMB/DMC Charter: ICH E6(R3), finalised by EMA in 2023, specifically requires a pre-specified Independent Data Monitoring Committee (IDMC) charter for Phase I trials involving novel mechanisms, dose escalation in vulnerable populations, or combination regimens.
Risk Management and Stopping Rules: A site-level and sponsor-level stopping rule document must be included. EMA inspectors at Phase I units assess whether these are operationalised in the study procedures.
The CMS assessment of Phase I CTAs under CTR 536/2014 typically runs 30 days for Part I (scientific review), with Concerned Member States having 10 additional days for Part II national compliance review. EMA’s 2024 CTIS performance data indicated a median CTA approval time of 41 days for Phase I oncology studies across five or more member states.
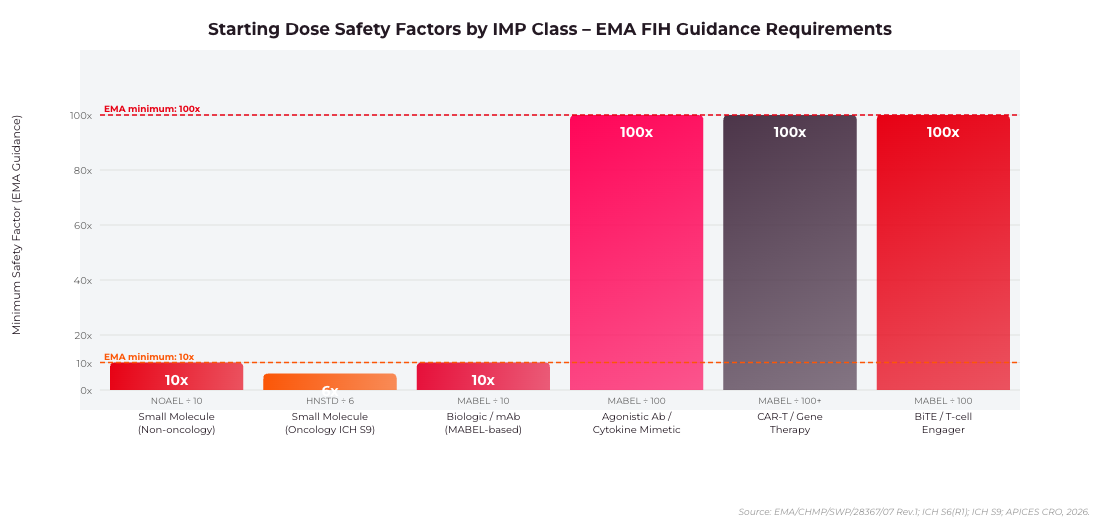
Figure 2: Minimum starting dose safety factors by IMP class as required by EMA/CHMP/SWP/28367/07 Rev.1. Agonistic antibodies, BiTEs and cell therapies require a minimum 100x safety factor over MABEL.
Starting Dose Selection: MABEL and NOAEL Approaches
MABEL for Biological IMPs
The MABEL approach – required by EMA for biologics, monoclonal antibodies, immunomodulatory agents, and any IMP with a novel mechanism – derives the starting dose from the lowest dose producing a measurable pharmacological effect in the most sensitive and relevant in vitro or in vivo system. The MABEL is then translated to human equivalent dose (HED) using body surface area (BSA) or body weight scaling, with an additional safety factor of 10 or higher depending on the steepness of the dose-response curve.
For IMPs with steep dose-response curves – including agonistic antibodies, cytokine-like molecules, and CAR-T cell therapies – EMA expects the starting dose to be at least 100-fold below the pharmacologically active dose in the most sensitive species. The TGN1412 incident in 2006 directly shaped this requirement, and subsequent EMA guidance has progressively tightened the justification standards expected from CTA applicants.
NOAEL-Based Derivation for Small Molecules
For small-molecule IMPs, the starting dose is typically derived from the NOAEL observed in the most sensitive species in GLP toxicology studies, with a safety factor of 10 applied as a standard minimum. For oncology indications where patients have exhausted standard-of-care options, ICH S9 allows a less conservative approach – HNSTD (Highest Non-Severely Toxic Dose) in rodents or NOAEL in non-rodents, with a safety factor of 6 for oncology FIH studies.
Where allometric scaling is used, the conversion factors in FDA’s 2005 guidance (retained as a regulatory reference by EMA) apply: a factor of 37 from mouse, 6.2 from monkey, and 3.1 from dog to human, based on BSA normalisation.
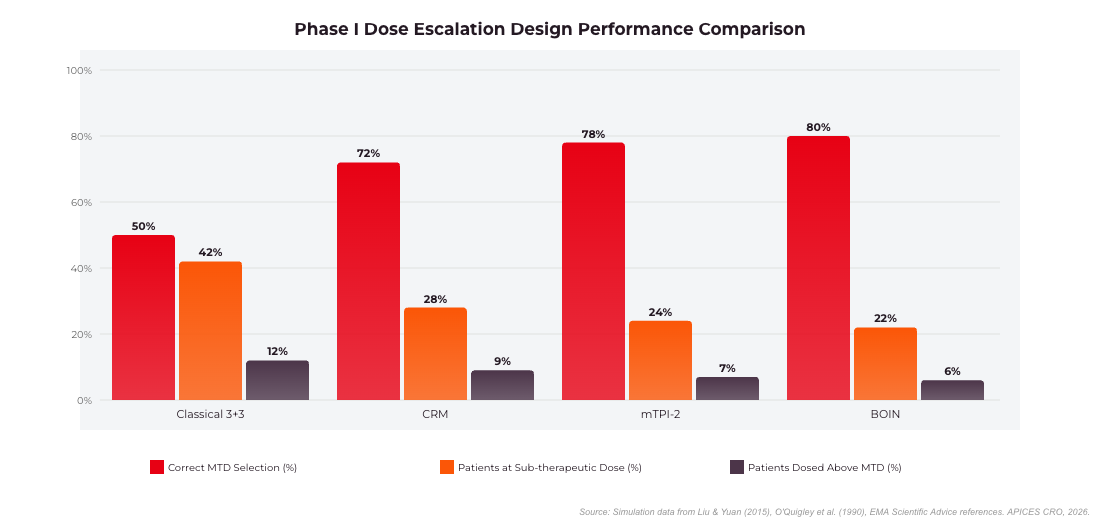
Figure 3: Phase I Dose Escalation Design Performance Comparison. BOIN and mTPI-2 achieve 80% correct MTD selection vs 50% for the classical 3+3 design. Source: Simulation data from Liu & Yuan (2015), O’Quigley et al. (1990).
Dose Escalation Design Options: Rule-Based vs. Model-Based
The Classical 3+3 Design
The 3+3 design has been the default for Phase I dose escalation for decades: three patients are enrolled at a dose level; if zero DLTs occur, escalation proceeds; if one DLT is observed, three additional patients are enrolled; if two or more DLTs occur at any level, escalation stops and the prior level is declared the Maximum Tolerated Dose (MTD). Despite its simplicity, the 3+3 design has well-documented limitations: it is statistically inefficient, frequently underestimates the true MTD, and tends to treat a higher proportion of patients at sub-therapeutic doses. Simulation studies have consistently shown that 3+3 correctly identifies the true MTD only 40–60% of the time under typical assumptions.
EMA has not prohibited the 3+3 design, but the CHMP Oncology Working Party has signalled in multiple Scientific Advice letters that model-based designs are preferred for FIH oncology studies, particularly for IMPs with a narrow therapeutic index or complex pharmacodynamic profiles.
Continual Reassessment Method (CRM)
The CRM, introduced by O’Quigley et al. in 1990, uses a Bayesian dose-toxicity model to update DLT probabilities after each cohort and identify the dose level with a DLT probability closest to a pre-specified target (typically 20–33%). CRM requires prior specification of the dose-toxicity skeleton – the assumed probabilities of DLT at each dose level – and is sensitive to prior misspecification. EMA has accepted CRM-based dose escalation in FIH studies for oncology biologics since approximately 2012, provided the protocol includes a full description of the prior, the likelihood model, and a simulation report demonstrating operating characteristics.
BOIN and mTPI-2: Current Best Practice
The Bayesian Optimal Interval (BOIN) design, developed by Liu and Yuan (2015), and its successor the modified Toxicity Probability Interval 2 (mTPI-2) design are now the most widely adopted model-based Phase I designs in oncology and are increasingly applied in non-oncology indications as well. Both designs use simple decision tables derived from a target DLT rate, making them easier to implement and audit than full CRM implementations.
BOIN requires specification of three parameters: the target DLT rate (phi, typically 0.25–0.30), and the lower and upper equivalence bounds (typically phi1 = 0.6 x phi, phi2 = 1.4 x phi). After each cohort, the observed DLT rate determines whether the dose should be escalated, retained, or de-escalated – without requiring a real-time statistician for intra-cohort decisions. The FDA issued a Project Optimus guidance update in 2023 specifically referencing BOIN as an acceptable design for oncology dose optimisation, and EMA Scientific Advice responses in 2024 have echoed this position. BOIN software is publicly available via the BOIN R package (CRAN) and a validated web application at www.trialdesign.org.
Accelerated Titration Designs
Accelerated Titration Designs (ATDs), introduced by Simon et al. in 1997, allow single-patient cohorts during initial dose escalation at low doses (typically below 1/3 of the expected MTD), switching to standard 3+3 or model-based cohorts once the first DLT or two instances of grade 2 toxicity are observed. ATDs reduce total patient exposure at sub-therapeutic doses and are compatible with BOIN or CRM after the switch point. EMA has accepted ATD hybrids in FIH oncology CTAs, provided the protocol specifies objective criteria for switching from single-patient to multi-patient cohorts.
Dose-Limiting Toxicity Definition and DLT Window
DLT Criteria Specification
The definition of a DLT is one of the most scrutinised elements of a Phase I protocol by EMA and national CMS reviewers. CTCAE v5.0 grading is the standard reference; the protocol must specify which grade 3 and grade 4 events are considered DLTs, which are excluded (e.g., grade 3 alopecia, expected tumour flare), and how non-haematological and haematological DLTs are weighted. For immunotherapy programmes, the protocol must address immune-related adverse events separately, as grade 3 irAEs may not always represent a dose-limiting phenomenon under the classical mechanistic framework.
For non-oncology Phase I studies – increasingly common in cardiovascular, neurological, and metabolic disease programmes – the DLT definition must account for the healthy volunteer population and the typically tighter safety margins expected by EMA. The ICH E14/S7B guidance update (2022) on QTc prolongation is directly relevant for Phase I cardiovascular safety assessments and should be cross-referenced in the DLT criteria for relevant IMPs.
DLT Observation Window
The DLT observation window is the period during which a toxicity event must occur to qualify as a DLT for escalation decision purposes. For cytotoxic oncology agents, a 21–28 day window aligned with the treatment cycle is standard. For biologics with long half-lives, immunomodulatory agents, or cell therapies, EMA expects the DLT window to be extended to at least 42 days or two half-lives of the IMP – whichever is longer – to capture delayed immune-mediated toxicities. Gene therapy IMPs may require even longer observation windows, and the DSMB charter must specify how late-onset events are handled after the formal dose-escalation phase.
Sentinel Dosing and Inter-Cohort Pause Requirements
Sentinel Dosing
EMA guidance EMA/CHMP/SWP/28367/07 Rev.1 recommends sentinel dosing for the first dose level of FIH studies with novel mechanisms or high-risk pharmacology. Sentinel dosing typically means treating one or two patients at the starting dose before enrolling the remainder of the cohort, with an observation period of 24–72 hours before the remaining cohort patients receive the IMP. This approach is particularly important for agonistic antibodies, cytokine mimetics, bispecific T-cell engagers (BiTEs), and CAR-T therapies, where cytokine release syndrome can occur rapidly.
CTIS protocol submissions for FIH studies involving these IMP classes should explicitly state the sentinel dosing plan, including the number of sentinel patients, the observation period, the monitoring requirements (vital signs frequency, cytokine panel if applicable), and the criteria for proceeding to full cohort dosing.
Inter-Cohort Review and DSMB Pause
The inter-cohort pause – the period between the last patient dosed in a cohort and the first patient dosed in the next dose level – must be sufficient to observe the full DLT window for at least the majority of patients in the prior cohort. EMA reviewers have flagged protocols where the inter-cohort pause was shorter than the DLT window, particularly in accelerated designs. The DSMB/DMC must review all safety data during this pause and issue a written recommendation before escalation proceeds, with documentation requirements as specified in ICH E6(R3).
EMA Scientific Advice for Phase I Dose Escalation Design
EMA Scientific Advice (SA) is the primary mechanism through which sponsors can obtain binding regulatory input on Phase I dose escalation design before CTA submission. The SA procedure is governed by EMA/CHMP/SAWP/72894/2008 Rev.1. For a Phase I FIH study, the typical SA briefing document should address: MABEL/NOAEL calculation and starting dose justification, dose escalation design choice and simulation results, DLT definition and window rationale, proposed MTD/RP2D definition, and stopping rule philosophy.
EMA SA responses for Phase I dose escalation questions are typically issued within 70–90 calendar days of the SA request validation. The cost of a full SA procedure is approximately EUR 75,000–EUR 95,000 for a standard procedure (2025 EMA fee schedule), though SME-eligible companies receive significant reductions. Parallel EMA-FDA Scientific Advice (Type C meetings on the FDA side) can be requested for transatlantic development programmes and reduces the risk of divergent design requirements at the IND/CTA stage.
Frequently Asked Questions
What is the difference between MABEL and NOAEL in EU Phase I dose escalation?
MABEL (Minimum Anticipated Biological Effect Level) is derived from the lowest pharmacologically active dose in the most sensitive in vitro or in vivo system, translated to a human equivalent dose with a conservative safety factor. It is required by EMA for biological IMPs, immunomodulatory agents, and any IMP where the mechanism could trigger rapid or severe immune responses. NOAEL (No Observed Adverse Effect Level) is derived from GLP toxicology studies in animals and is the standard starting point for small-molecule IMPs without high-risk pharmacological mechanisms. For oncology small molecules, ICH S9 permits the HNSTD-based approach, which is less conservative than NOAEL. EMA requires sponsors to calculate both MABEL and NOAEL when applicable and use the more conservative result as the starting dose.
Is the 3+3 dose escalation design still acceptable to EMA in 2025?
Yes, EMA has not prohibited the 3+3 design. However, EMA’s CHMP Oncology Working Party has consistently recommended model-based designs (BOIN, mTPI-2, CRM) in Scientific Advice responses for oncology FIH studies, citing the higher statistical efficiency and more accurate MTD identification of these methods. For non-oncology Phase I studies in healthy volunteers where the therapeutic index is wide and the starting dose is very conservative, 3+3 may still be accepted without challenge. Sponsors electing to use 3+3 for oncology FIH studies should be prepared to justify this choice explicitly in the protocol and anticipate CHMP questions during CTA review.
What documents must a CTIS Phase I CTA submission include for dose escalation?
A CTIS Part I CTA submission for a Phase I dose escalation study must include: a protocol with the full escalation algorithm, DLT definitions, DLT window, and stopping rules; an Investigator’s Brochure with MABEL or NOAEL justification for starting dose; a non-clinical study report summary addressing pharmacokinetics and toxicology; and, if a model-based design is used, a simulation report demonstrating the design’s operating characteristics (probability of correct MTD selection, proportion of patients treated above the true MTD). The DSMB charter is required under ICH E6(R3) and must be included in the CTA package or provided as a condition of approval depending on the CMS.
How long does EMA take to authorise a Phase I CTA under CTR 536/2014?
Under CTR 536/2014, the CMS has 30 calendar days to complete the Part I scientific assessment of a Phase I CTA, with a 10-day clock-stop permitted for sponsor responses to list-of-questions. Part II national compliance assessment runs in parallel and takes up to 30 days for the member state. EMA’s 2024 CTIS performance report indicated a median total authorisation time (from validation to authorisation) of 41 days for multi-country Phase I oncology studies and 38 days for non-oncology Phase I studies, assuming no clock-stops. Submissions with incomplete dose escalation documentation or missing DLT window justification are the most common reason for list-of-questions in CHMP CMS reviews.
Can BOIN dose escalation be used for non-oncology Phase I studies in the EU?
Yes. While BOIN was developed primarily in the oncology context, its underlying statistical framework – Bayesian optimal interval decision rules based on a target DLT rate – is applicable to any Phase I dose escalation study where a DLT can be operationally defined. EMA has accepted BOIN in non-oncology Phase I study protocols reviewed through Scientific Advice, including cardiovascular, CNS, and infectious disease programmes. The key requirement is that the target DLT rate, equivalence interval bounds, and simulation operating characteristics are documented in the protocol and, where appropriate, in the CTA briefing document. For healthy volunteer Phase I studies, a target DLT rate of 0.20–0.25 is typical, compared to 0.25–0.33 in oncology studies.
Selecting the right phase I dose escalation design is one of the most consequential decisions in a drug development programme – it determines the efficiency of safety characterisation, the probability of correctly identifying the Maximum Tolerated Dose or Recommended Phase 2 Dose, and the regulatory defensibility of your transition to Phase II. APICES CRO’s early clinical development team has supported Phase I CTA submissions across more than 12 EU member states, providing dose escalation design selection, simulation packages, DSMB charter development, and MABEL/NOAEL documentation for both oncology and non-oncology programmes. Contact APICES CRO to discuss your Phase I programme and how we can support your EU regulatory strategy from IND/CTA design through to RP2D selection.
Get the latest clinical insights delivered to your feed or inbox!
Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I...
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
Complete guide to Phase I oncology trials in Western Europe in 2026: CTIS submissions, country selection (Spain, France, Germany, Netherlands), site criteria, study design, and CRO selection for biotech sponsors.
")
")