This guide explores how clinical trials are conducted in Belgium, covering the regulatory framework, ethics review process, site infrastructure, and why Belgium stands out as a specialist hub for oncology and rare disease research in Western Europe.
Clinical Trials in Belgium: A Specialist Hub for Oncology and Rare Diseases in Western Europe
Belgium occupies a distinctive position in European clinical research. Despite its relatively small population of approximately 11.6 million, the country hosts some of the continent’s most respected academic medical centres and has developed a regulatory environment that, under the EU Clinical Trials Regulation (EU CTR, Regulation EU 536/2014), consistently delivers competitive approval timelines. Belgium functions most effectively as a high-quality supplementary country in multinational programmes rather than as a standalone enrolment engine.
The country’s clinical trial infrastructure is concentrated in university hospitals in Brussels, Leuven, Ghent, Antwerp, and Liège. These institutions have deep experience in early-phase studies, particularly in oncology and rare diseases, and maintain long-standing relationships with international sponsors. Belgium’s research community is internationally oriented, with investigators routinely publishing in leading journals and participating in global trial networks.
Sponsors should be aware of Belgium’s bilingual complexity: sites in Brussels operate in both French and Dutch, Wallonia predominantly in French, and Flanders in Dutch. Patient-facing documents – informed consent forms, patient diaries, recruitment materials – must be translated accordingly, which adds both cost and coordination overhead. For sponsors already running EU programmes, Belgium typically integrates smoothly via CTIS.
Quick Facts
Metric
Belgium
Competent Authority
FAMHP (Federal Agency for Medicines and Health Products)
Ethics Review
Accredited Lead Ethics Committee (CEC model)
Approval Timeline
~45–55 days (FAMHP + parallel ethics)
Population
~11.6 million
Strengths
Oncology, rare diseases, neurology, immunology
Why Sponsors Choose Belgium for Clinical Trials
Belgium’s appeal rests on the quality of its academic infrastructure and the international orientation of its investigators. Several factors make it a consistent choice for sponsors adding a Western European site to a multinational programme:
World-class academic hospitals with dedicated early-phase and Phase I/II units
Investigators experienced in multinational trials and GCP-compliant documentation
Competitive approval timelines under the EU CTR framework
Central geographic location within the EU, facilitating sponsor oversight visits
Strong biobank and translational research capabilities, particularly at KU Leuven and VUB
That said, Belgium is not the right primary enrolment country for large Phase III trials with broad inclusion criteria. Its patient pool is limited, and per-patient costs are among the higher end in the EU. Sponsors should plan Belgium as a quality contributor rather than a volume contributor.
Regulatory Framework for Clinical Research in Belgium
Belgium fully implements the EU Clinical Trials Regulation (EU CTR, Regulation EU 536/2014) and submits applications through the Clinical Trials Information System (CTIS). The competent authority is the Federal Agency for Medicines and Health Products (FAMHP), which handles regulatory assessment in parallel with the ethics review conducted by an accredited lead ethics committee under the Centralised Ethics Committee (CEC) model.
Belgium adopted the CEC model under the EU CTR, which means a single lead ethics committee conducts the Part II review on behalf of all Belgian sites. This significantly simplifies the ethics review process compared to older national frameworks, where sponsors previously needed separate ethics approvals from each site’s local committee. The consolidation has shortened timelines and reduced administrative duplication.
For non-IMP studies, observational research, and post-authorisation studies, separate national legislation applies. Sponsors running non-interventional programmes should consult Belgian requirements separately from the EU CTR pathway.
Clinical Trial Approval Process in Belgium (Step-by-Step)
Step 1 – CTIS submission: Sponsor submits the application via CTIS. Belgium is selected as a Member State Concerned (MSC) if not acting as the Reporting Member State.
Step 2 – Validation: CTIS validates the submission for completeness. Incomplete dossiers are returned promptly; sponsors should ensure all required documentation is in order before submission.
Step 3 – Part I assessment (FAMHP): FAMHP conducts the scientific and safety review. This runs in parallel with the Part II ethics review.
Step 4 – Part II assessment (Lead Ethics Committee): The accredited lead ethics committee reviews the ethical aspects of the trial, including the informed consent forms in French and Dutch.
Step 5 – Request for information (RFI): Either FAMHP or the ethics committee may issue an RFI. The clock stops during the sponsor’s response period.
Step 6 – Decision: FAMHP issues the authorisation decision via CTIS. Site-level contracts and local feasibility checks can proceed in parallel.
Clinical Trial Approval Timeline in Belgium (How Long It Takes)
Under the EU CTR, Belgium typically achieves regulatory and ethics decisions within the statutory assessment windows. In practice, sponsors should plan for the following indicative timelines, assuming a complete and well-prepared dossier:
Step
Estimated Timeline
CTIS submission and validation
1–5 working days
FAMHP Part I assessment
~30–45 days
Lead Ethics Committee Part II assessment
~30–45 days (parallel)
RFI response period (if applicable)
12–31 days (clock stopped)
Site contract and budget negotiation
4–12 weeks (site dependent)
Estimated total from submission to first patient
~3–5 months
Belgium is one of the more predictable EU countries for timeline planning. RFIs are common and should be anticipated. Sites with active clinical research units – particularly UZ Leuven and Jules Bordet – tend to move through internal contract processes more quickly than smaller hospitals.
Clinical Trial Sites in Belgium
Belgium’s principal clinical trial sites are concentrated in the major university hospital networks. Key institutions include:
Jules Bordet Institute, Brussels – National cancer centre with Phase I/II units; one of Belgium’s most active oncology trial sites
UZ Leuven (Universitair Ziekenhuis Leuven) – Flagship academic hospital linked to KU Leuven; strong across oncology, haematology, rare diseases, and transplantation
UZ Gent (Universitair Ziekenhuis Gent) – Major Flemish academic hospital; active in respiratory, oncology, and nephrology
Cliniques Saint-Luc, Brussels – French-speaking university hospital linked to UCLouvain; strong in oncology, hepatology, and neurology
UZA (Universitair Ziekenhuis Antwerpen) – Antwerp university hospital; strong in rare diseases, metabolic disorders, and haematology
CHU de Liège – Principal academic hospital for the Liège region; active in haematology, oncology, and gastroenterology
Site selection in Belgium requires careful attention to language. Investigators and coordinators at most sites are proficient in English, but patient-facing materials must be available in French and/or Dutch depending on the site’s location. Budget for dual-language translation from the outset.
Patient Recruitment in Belgium
Belgium’s patient pool is inherently limited by its population size. For common oncology indications, Belgium can contribute meaningfully as one site among several in a multinational programme, but sponsors should not rely on it as a primary enrolment country for trials requiring large sample sizes. Rare disease trials benefit from Belgium’s strong patient registries and specialist centres, which can identify eligible patients across the country relatively efficiently.
Recruitment is supported by the Belgian national health insurance system (RIZIV/INAMI), which provides relatively uniform access to healthcare, reducing socioeconomic barriers to trial participation. University hospitals maintain active patient advocacy relationships and disease-specific registries that aid screening. However, competition for patients at the major academic sites is high, as Belgium’s leading hospitals participate in many simultaneous international trials.
Therapeutic Areas with Strong Clinical Trial Activity in Belgium
Oncology and haematology – Belgium’s dominant area; Jules Bordet, UZ Leuven, and multiple sites run early and late-phase oncology trials
Rare diseases – Strong national centres of expertise (Centra voor Zeldzame Aandoeningen) with European Reference Network (ERN) affiliations
Neurology and neurodegenerative diseases – Active programmes at UZ Leuven and UZ Gent
Immunology and rheumatology – Strong research output and patient populations at major academic centres
Cardiovascular and metabolic diseases – Active programmes, though less dominant than oncology
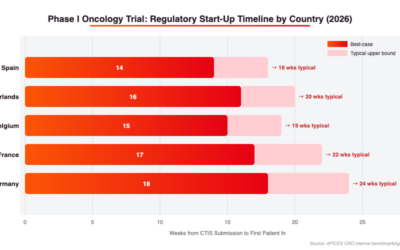
Clinical Trial Start-Up Timelines in Belgium
Start-up in Belgium is generally predictable but not the fastest in Europe. Internal hospital contracting processes can add time beyond the regulatory approval window, particularly at sites that require review by multiple internal committees. The following represents typical ranges:
Activity
Estimated Duration
Regulatory and ethics approval (CTIS)
45–55 days
Site contract and budget negotiation
6–12 weeks
Site initiation visit (SIV) and staff training
2–4 weeks
IRB/IEC local formalities (site-level)
2–4 weeks
First patient screened
~4–6 months from submission
Sponsors with existing site relationships in Belgium typically experience faster contracting. It is worth investing in feasibility and site engagement before submission to compress the post-approval phase.
Costs of Conducting a Clinical Trial in Belgium
Belgium sits in the moderate-to-high cost range for European clinical research. Healthcare and research staff salaries are higher than in Central and Eastern Europe, and hospital overhead charges at academic sites can be substantial. Per-patient costs for a Phase II oncology study typically range from €18,000 to €40,000, depending on protocol complexity, visit schedule, and the specific site.
Key cost drivers include: dual-language document translation, site management fees at academic hospitals, and the cost of specimen handling and centralised laboratory logistics. Belgium’s national healthcare system does cover standard-of-care costs for trial participants in most cases, which partially offsets the per-patient charge to sponsors.
When Belgium Is the Right Choice for Clinical Trials
Belgium is particularly well suited to trials where investigator expertise and site quality are paramount over enrolment volume. It is an appropriate choice when:
The indication is rare or highly specialised, and Belgium’s reference centres hold relevant patient registries
Phase I or early Phase II studies require experienced investigators and well-equipped research units
The sponsor is running an EU-wide programme and wants a high-quality Western European site alongside France, Germany, or the Netherlands
Translational research components (biomarkers, tissue banking, imaging sub-studies) benefit from Belgium’s academic infrastructure
Belgium is less suitable as a standalone or primary enrolment country for Phase III trials with large sample size requirements, or for sponsors with very tight budgets.
Advantages of Conducting Clinical Studies in Belgium
Full CTIS integration under EU CTR simplifies multi-country European submissions
Consolidated ethics review under CEC model reduces administrative complexity
Internationally oriented investigators with strong publication records and trial experience
Excellent oncology and rare disease centres with ERN affiliations
Central EU location for sponsor oversight and monitoring visits
Strong biobank and translational research infrastructure
How APICES Supports Clinical Trials in Belgium
APICES provides full-service CRO support for clinical trials in Belgium, including regulatory submissions via CTIS, ethics committee liaison, site identification and feasibility, site management, and patient recruitment strategy. Our team has established relationships with the principal academic sites and understands the bilingual requirements that affect patient-facing document preparation.
We support sponsors from protocol development and CTIS submission through to site close-out and final reporting, managing the French/Dutch language requirements and coordinating with FAMHP and the lead ethics committee throughout the trial lifecycle. For sponsors incorporating Belgium into a broader European programme, we provide integrated oversight across all sites and member states.
Frequently Asked Questions About Clinical Research in Belgium
How long does clinical trial approval take in Belgium? Under the EU CTR, Belgium processes applications through CTIS with the FAMHP conducting the Part I (scientific and safety) assessment and an accredited lead ethics committee conducting the Part II (ethical) review simultaneously. For a complete and well-prepared dossier, the combined timeline is approximately 45–55 days from validation. Requests for information (RFIs) from either the FAMHP or the ethics committee will pause the statutory clock, so sponsors should ensure their dossiers are thorough before submission to minimise RFI risk.
Does Belgium use CTIS for clinical trial applications? Yes. Belgium has fully adopted the EU Clinical Trials Regulation (EU CTR, Regulation EU 536/2014) and all interventional clinical trial applications are submitted through the Clinical Trials Information System (CTIS). The Federal Agency for Medicines and Health Products (FAMHP) is Belgium’s national competent authority. Belgium can act as either the Reporting Member State (RMS) or a Member State Concerned (MSC) in multi-country applications.
What languages are required for patient documents in Belgium? Belgium’s linguistic geography requires careful planning. French is required at Wallonian sites and most Brussels institutions; Dutch is required at Flemish sites and also at many Brussels institutions. Sites in the Brussels Capital Region typically request both French and Dutch versions of all patient-facing materials, including informed consent forms, patient information leaflets, and any diaries or questionnaires. German is required for the small German-speaking community in the east of the country. Research staff and investigators routinely communicate in English with international sponsors.
What are the main clinical trial sites in Belgium? Belgium’s principal clinical trial sites are its major university hospital networks: Jules Bordet Institute in Brussels (national cancer centre and leading oncology trial site), UZ Leuven (linked to KU Leuven; strong in oncology, haematology, and rare diseases), UZ Gent (strong in respiratory and nephrology), Cliniques Saint-Luc in Brussels (linked to UCLouvain), UZA in Antwerp (strong in rare diseases and metabolic disorders), and CHU de Liège (haematology and gastroenterology). Smaller regional hospitals may be relevant for specific indications.
Is Belgium suitable for rare disease clinical trials? Belgium is one of the stronger European countries for rare disease research. The national centres of expertise (Centra voor Zeldzame Aandoeningen) at UZ Leuven, UZA Antwerp, and other institutions hold ERN affiliations and maintain disease-specific registries that can be used to identify eligible patients. For pan-European rare disease programmes, Belgium’s ERN-affiliated centres are natural partners. The relatively small national population is partially offset by the depth of the specialist registries and the international referral networks of the academic centres.
What does a Phase II oncology trial cost per patient in Belgium? Per-patient costs for a Phase II oncology trial in Belgium typically range from €18,000 to €40,000. The wide range reflects differences in protocol complexity (number of visits, imaging, laboratory assessments, tissue processing), site overhead charges, and whether the trial involves novel therapeutics requiring specialist handling. Belgium’s national health insurance system (RIZIV/INAMI) covers standard-of-care elements for trial participants in most cases, which partially reduces the per-patient charge to sponsors. Sponsors should budget separately for dual-language document translation and Belgian-specific regulatory fees.
Get the latest clinical insights delivered to your feed or inbox!
Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I...
Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based...
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
")
")