First-in-Human Trials in the EU: Regulatory Requirements and Site Selection in 2026
First-in-Human Trials in the EU: Regulatory Requirements and Site Selection in 2026 Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I (scientific/safety) and Part II […]
First-in-Human Trials in the EU: Regulatory Requirements and Site Selection in 2026
Key Takeaways: First-in-human trials in the EU are governed by Regulation (EU) No 536/2014 and the EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1). Sponsors must submit a complete Clinical Trial Application via the CTIS portal and obtain both Part I (scientific/safety) and Part II (ethics) authorisation before first patient dosing. Starting dose selection must be justified using MABEL and/or NOAEL methodology, with the lower result selected where both apply. Country-specific CTA timelines vary significantly: Spain and Belgium consistently achieve authorisation within 35–50 days, while Germany can require 60–90 days. A well-prepared IMPD, sentinel dosing provisions, and pre-submission engagement with the relevant NCA substantially reduce clock-stop risk.
First-in-human trials in the EU represent the critical bridge between promising preclinical data and the first demonstration of tolerability and pharmacokinetics in human subjects. For sponsors operating under Regulation (EU) No 536/2014, every procedural decision – from CTIS submission strategy to Phase I unit selection – carries regulatory and operational consequences that can add months to development timelines. This article sets out the current EU framework for first-in-human trials, starting dose methodology, site qualification standards, and country-specific considerations that inform the design and execution of a competitive FIH programme in 2025.
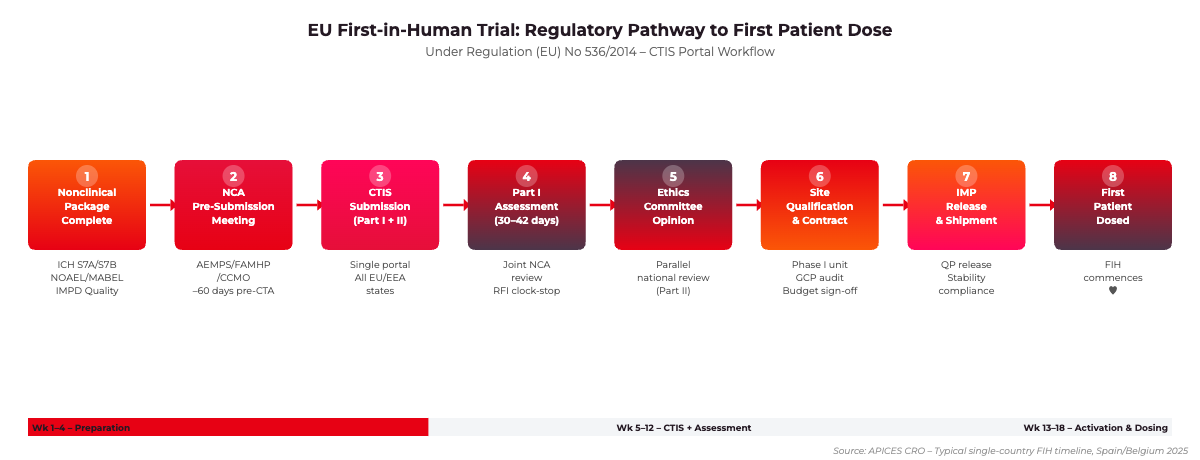
EU FIH Regulatory Pathway: From Nonclinical Package Completion to First Patient Dose. Source: APICES CRO, 2025.
The EU Regulatory Framework for First-in-Human Trials
Regulation (EU) No 536/2014, which entered full application on 31 January 2022 via the CTIS portal, replaced Directive 2001/20/EC and unified the Clinical Trial Application process across all EU/EEA Member States. For FIH sponsors, the most consequential change is the dual Part I/Part II dossier structure. Part I contains the scientific and safety documentation assessed jointly by the Member States concerned, while Part II contains national ethics committee documentation assessed independently by each country.
The CTIS Submission Workflow
Under the current framework, the sponsor submits all CTA components simultaneously through the CTIS portal. The regulatory clock for Part I runs for 30 days from validation, with a 12-day extension permitted for complex applications – a category that almost always applies to FIH trials involving novel mechanisms of action. Member States may raise requests for information (RFI) during this period, which triggers a clock-stop and pauses assessment until the sponsor responds. In practice, the total calendar time from CTIS submission to first authorisation in a multinational FIH trial ranges from 45 to 90 days depending on the countries selected and the quality of the initial dossier.
Sponsors should engage the relevant National Competent Authority in pre-submission scientific advice at least 60 days before CTIS submission. AEMPS in Spain, FAMHP in Belgium, and the CCMO in the Netherlands each offer formal pre-submission meetings specifically adapted to FIH applications. This engagement substantially reduces the probability of a clock-stop related to starting dose justification or nonclinical data package completeness.
The IMPD is the scientific centrepiece of the Part I dossier. For FIH applications, the IMPD must contain four modules: Quality (CMC), Nonclinical, Clinical (prior human exposure if any), and a Risk/Benefit assessment. The EMA’s Quality of Medicines group expects at minimum a characterised drug substance with established specifications, a manufacturing process description, and stability data covering the proposed clinical shelf-life. For biologics, requirements align with ICH Q6B. The nonclinical module must demonstrate safety pharmacology data per ICH S7A and S7B, repeat-dose toxicity data of sufficient duration relative to the proposed dosing schedule, and genotoxicity data for small molecules. For oncology FIH programmes, ICH S9 provides a modified nonclinical package with streamlined reproductive toxicity requirements.
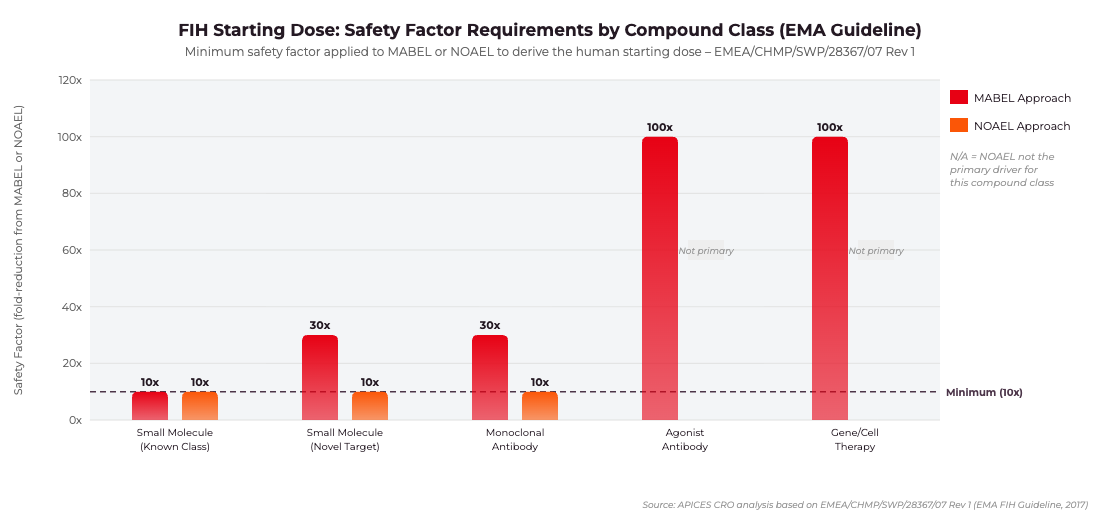
FIH Starting Dose Safety Factor Requirements by Compound Class. Based on EMEA/CHMP/SWP/28367/07 Rev 1. Source: APICES CRO analysis, 2025.
Starting Dose Selection: MABEL, NOAEL, and the EMA First-in-Human Guideline
The EMA’s guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products (EMEA/CHMP/SWP/28367/07 Rev 1, 2017) is the primary reference for starting dose determination in the EU. The guideline does not mandate a single methodology but requires sponsors to apply the most appropriate approach based on the mechanism of action and the available nonclinical dataset. Where both MABEL and NOAEL are calculated, the lower resulting human dose must be selected.
MABEL-Based Starting Dose
The Minimum Anticipated Biological Effect Level (MABEL) approach is mandatory for high-risk agents: those with novel mechanisms of action, narrow therapeutic windows, immunomodulatory activity, or potent agonist activity on immune cells. MABEL is derived from the entire in vitro and in vivo pharmacological dataset and represents the dose expected to produce the minimum pharmacodynamic effect in humans. Sponsors must integrate data from receptor binding studies, in vitro cell-based assays, and animal pharmacodynamic models, applying appropriate safety factors – typically between 10-fold and 100-fold – to arrive at the clinical starting dose. The 2006 TGN1412 incident, in which six healthy volunteers suffered life-threatening cytokine release following a CD28 superagonist antibody dose calculated solely on the basis of animal NOAEL, directly motivated the EMA’s adoption of MABEL as the primary tool for high-risk FIH dose selection.
NOAEL-Based Starting Dose
For compounds with established mechanisms of action and a well-characterised nonclinical safety profile, the No Observed Adverse Effect Level approach remains acceptable. The human equivalent dose is calculated from the NOAEL in the most sensitive relevant species using body surface area normalisation and a safety factor of at least 10. Where toxicity is steep or species differences are uncertain, a higher safety factor is required. The EMA guideline explicitly states that where MABEL and NOAEL give different starting doses, the lower dose must be selected. This requirement – frequently overlooked by sponsors accustomed to FDA submissions where NOAEL is often the primary driver – has been the source of numerous clock-stop requests from European NCAs in 2023–2025.
FIH Trial Design Requirements for EU Regulatory Acceptability
The design of the FIH protocol must satisfy both scientific rigour and EU regulatory expectations on participant protection. The EMA is particularly attentive to sentinel dosing provisions, dose-escalation decision criteria, stopping rules, and DSMB composition and independence.
Dose Escalation Schema
Traditional 3+3 escalation designs are increasingly regarded as inefficient and are being superseded in EMA-reviewed protocols by model-informed designs. The three most frequently used in current EU FIH submissions are:
mTPI-2 (Modified Toxicity Probability Interval): A Bayesian adaptive design that uses dose-toxicity intervals rather than point estimates. The EMA expects the operating characteristics table demonstrating DLT probability intervals and escalation/de-escalation rules to be included in the protocol appendix. mTPI-2 is accepted for both oncology and non-oncology FIH programmes.
BOIN (Bayesian Optimal Interval): A two-parameter Bayesian design operationally comparable to 3+3 but with optimised decision rules and superior operating characteristics at the true MTD. BOIN requires pre-specification of the target DLT rate and equivalence intervals, which must be documented in the statistical analysis plan.
Accelerated Titration Design (ATD): Acceptable for low-risk agents where the steep dose-toxicity relationship of standard designs would unnecessarily expose participants to sub-therapeutic doses. Single-patient cohorts are permissible until the first Grade 2 toxicity is observed, at which point standard multi-patient cohorts are required.
Sentinel Dosing and Observation Periods
For non-oncology FIH trials, the EMA strongly recommends sentinel dosing – typically one to two subjects receiving active drug before the remaining cohort members are dosed – with a minimum observation period of 24 hours before the cohort proceeds. For compounds with delayed mechanisms or extended half-lives, longer observation windows are expected. The protocol must specify exactly how sentinel data will be reviewed and by whom, and the DSMB charter must define the quorum, independence requirements, and decision-making process for dose escalation approval.
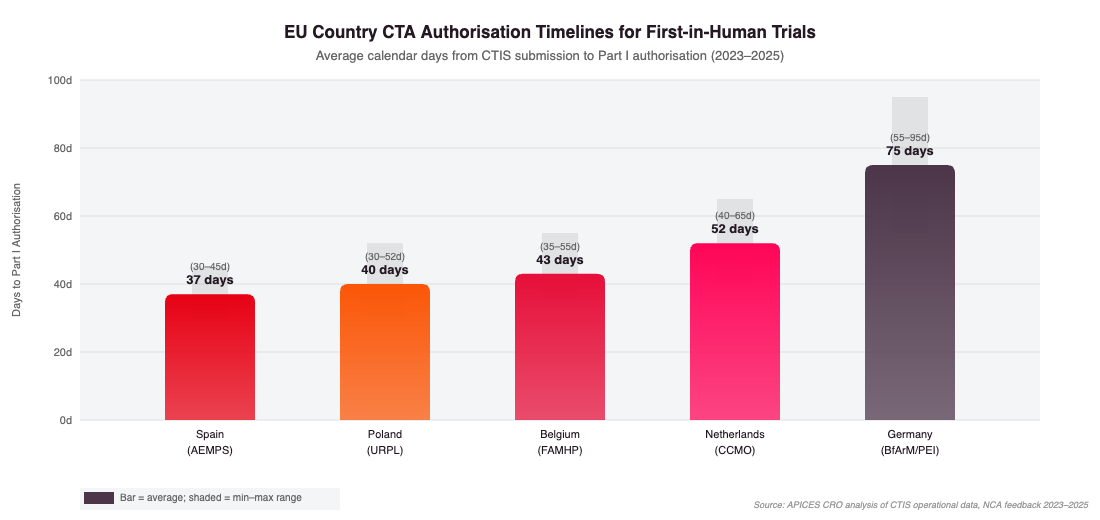
EU Country CTA Authorisation Timelines for FIH Trials. Average calendar days from CTIS submission to Part I authorisation. Source: APICES CRO analysis, 2023–2025.
Country Selection and Phase I Unit Standards in Europe
The selection of countries and Phase I units for first-in-human trials is a strategic decision with direct implications for timeline, patient population access, and regulatory complexity. Across the EU/EEA, Spain, Belgium, the Netherlands, and Poland represent the most competitive FIH locations for industry sponsors, offering a combination of fast NCA review timelines, experienced Phase I units, and competitive site costs.
Key EU Jurisdictions for First-in-Human Trials
Spain (AEMPS): Consistently the fastest EU NCA for FIH CTA authorisation, averaging 30–40 days for Part I approval. AEMPS offers a voluntary pre-submission meeting that significantly reduces clock-stop risk. Spain hosts START Madrid–CIOCC, CIMA (Centro de Investigación Médica Aplicada), and multiple AEEC-accredited Phase I units with combined capacity exceeding 200 Phase I beds.
Belgium (FAMHP): Averaging 35–50 days for Part I authorisation. Belgium’s single central ethics committee process is efficient and well-adapted to multinational trials. UZ Leuven Early Phase Oncology Unit, Institut Jules Bordet, and UZA Phase I programme represent leading options.
Netherlands (CCMO): Averaging 45–60 days. The CCMO provides phase-specific guidance documents for FIH applications and operates a highly transparent assessment process. AMC (Amsterdam UMC) and UMC Utrecht host established Phase I units with specialist PK/PD infrastructure.
Poland (URPL): Averaging 30–50 days with a highly experienced investigator base and site costs approximately 40–50% lower than Western Europe. Particularly advantageous for oncology FIH programmes requiring rapid parallel accrual across multiple cohorts.
Germany (BfArM/PEI): Averaging 60–90 days, with the Paul-Ehrlich-Institut applying particularly rigorous scrutiny to biological agents. Germany’s extended review timeline is partially offset by access to a large patient population and prestigious academic Phase I infrastructure through the DKTK Early Clinical Trials Network.
Phase I Unit Qualification Criteria
Sponsors should evaluate potential Phase I units against the following minimum qualification criteria before finalising site selection:
Dedicated Phase I beds with 24-hour medical coverage: The unit must have dedicated inpatient beds under direct Phase I supervision, separate from general ward capacity, with around-the-clock physician coverage by staff experienced in Phase I adverse event management.
Emergency response capability: Full resuscitation equipment, documented on-site ICU access or rapid transfer protocol, and tested response procedures for anaphylaxis, cytokine release syndrome, and cardiovascular emergencies.
Validated bioanalytical infrastructure: Either an on-site validated bioanalytical laboratory or documented agreements with a central laboratory, with turnaround time commitments compatible with PK-driven dose escalation decision timelines.
Clean GCP inspection history: Review the unit’s GCP inspection record with both the EMA and the national GCP inspectorate. Any Critical findings within the last five years require detailed explanation and corrective action evidence before site qualification can proceed.
Adequate throughput capacity: Confirm the unit’s current and projected trial load to ensure it can accommodate the proposed start date and escalation schedule without competing resource conflicts that could compromise observation period compliance.
Risk Categorisation and Specific Risk Mitigation Measures
The EMA’s FIH guideline introduces a risk categorisation framework that determines the intensity of risk mitigation required in the study protocol. High-risk compounds – including biological agents targeting immune function, novel agonists with potential for cytokine release, and compounds with demonstrated off-target binding at pharmacologically relevant concentrations – require a comprehensive specific risk mitigation package.
Required specific risk mitigation measures for high-risk first-in-human trials in the EU typically include: mandatory inpatient observation for at least 24 hours post-dose, predefined cytokine monitoring schedules with laboratory turnaround time commitments, immediate-access emergency medications pre-positioned at the bedside (corticosteroids, antihistamines, epinephrine), dose escalation decisions requiring independent DSMB review after every cohort, and pre-specified clinical halt criteria that automatically suspend dosing pending safety review. The DSMB charter must document the independence of all DSMB members from the sponsor and principal investigators and must define minimum response timelines for emergency safety reviews.
Practical Timeline Planning for EU FIH Programmes
Sponsors planning a first-in-human programme in the EU should construct their timeline from a realistic understanding of the sequential and parallel dependencies between regulatory, operational, and scientific workstreams. Based on CTIS operational experience from 2022–2025, the following milestones represent realistic planning assumptions for a single-country FIH trial in Spain or Belgium:
IMPD and protocol completion: 12–16 weeks before planned first-patient-in date
Pre-submission meeting with NCA: 8–10 weeks before CTIS submission
CTIS submission: 10–12 weeks before planned first-patient-in
Part I authorisation (Spain/Belgium): 5–7 weeks after submission
Ethics committee opinion: 5–8 weeks after submission (runs in parallel with Part I)
Site qualification and contract execution: 6–10 weeks (overlaps with regulatory review)
IMP release and shipment: 2–4 weeks after authorisation
First patient screened and dosed: 2–3 weeks after IMP arrival at site
Multinational FIH programmes add complexity at the Part II stage, where each Member State’s ethics process runs independently and on different timelines. Country selection should account for ethics committee review timelines alongside NCA review timelines. In practice, ethics committee delays – rather than NCA Part I delays – are the more common rate-limiting step in Spain and Belgium for well-prepared applications.
Frequently Asked Questions
What is the difference between a first-in-human trial and a Phase I trial in EU regulatory terminology?
A first-in-human trial is the initial administration of an investigational medicinal product to human subjects – typically healthy volunteers for non-oncology compounds, or patients for oncology agents where a healthy volunteer population is ethically inappropriate. Phase I is a broader clinical development phase that may include FIH trials but also encompasses early dose-ranging, PK/PD, drug-drug interaction, and preliminary efficacy studies. All FIH trials in the EU require a full CTA under Regulation (EU) No 536/2014, regardless of whether they carry the label Phase I, Phase Ia, or Phase 0.
How long does it take to obtain CTA authorisation for a first-in-human trial in the EU?
The formal Part I assessment clock under Regulation (EU) No 536/2014 runs for 30 days from CTIS validation (extendable to 42 days for complex applications). In practice, clock-stops triggered by NCA requests for information commonly extend total calendar time to 45–90 days. Spain and Belgium are the fastest jurisdictions, averaging 35–50 days for well-prepared FIH dossiers. Germany can require 60–90 days, particularly for biological agents subject to Paul-Ehrlich-Institut assessment.
Is MABEL or NOAEL the required methodology for EU first-in-human starting dose selection?
The EMA’s first-in-human guideline (EMEA/CHMP/SWP/28367/07 Rev 1) requires sponsors to evaluate both MABEL and NOAEL where applicable and to select the lower resulting starting dose. For high-risk agents – immunomodulators, agonist antibodies, compounds with steep dose-response relationships – MABEL is the primary methodology. For small molecules with well-understood mechanism classes, NOAEL may serve as the primary driver, but MABEL must still be assessed and documented in the IMPD nonclinical module. Failure to provide a complete MABEL analysis for a high-risk compound is one of the most common clock-stop triggers encountered in current CTIS applications.
Can a sponsor run a first-in-human trial across multiple EU countries simultaneously?
Yes. Regulation (EU) No 536/2014 was specifically designed to facilitate multinational clinical trials through the single CTIS portal. The Part I assessment is conducted jointly by a Reporting Member State and co-Reporting Member States, with a single Part I conclusion binding all concerned Member States. Part II ethics assessments run in parallel but independently in each country. All concerned Member States must issue both Part I and Part II authorisation before trial activities can commence in that specific country, so the operational start date in each country is determined by whichever authorisation is last issued.
What are the most common reasons for clock-stop in EU first-in-human CTA applications?
Based on CTIS operational data and NCA feedback through 2024–2025, the most frequent clock-stop triggers in FIH applications are: (1) insufficient MABEL analysis for high-risk agents; (2) incomplete nonclinical safety pharmacology data per ICH S7A or S7B; (3) IMPD quality module deficiencies including missing batch analysis data or inadequate stability data; (4) unclear stopping rules or an inadequate DSMB charter that fails to address independence requirements; and (5) inconsistencies between the protocol synopsis and the full protocol, which generate document concordance queries from the Reporting Member State. Proactive pre-submission engagement with the NCA is the most effective single measure for reducing clock-stop risk.
APICES CRO provides end-to-end support for first-in-human trials in the EU, from IMPD preparation and CTIS submission management to Phase I unit selection and on-site monitoring. Our senior clinical development consultants have managed FIH programmes across Spain, Belgium, the Netherlands, Poland, and Germany for biotech and pharmaceutical sponsors at every stage of development. To discuss your specific FIH programme requirements, visit our Early Clinical Development solutions page or contact our team for a confidential programme assessment.
Get the latest clinical insights delivered to your feed or inbox!
Key Takeaways: Phase I dose escalation design in the EU is governed by ICH E8(R1), EMA first-in-human guidance (EMA/CHMP/SWP/28367/07 Rev.1), and EU Clinical Trial Regulation 536/2014 via CTIS. The classical 3+3 design is increasingly replaced by model-based...
EU MDR clinical investigation requirements have fundamentally changed how Medtech sponsors plan and execute device studies in Europe. Regulation (EU) 2017/745 replaced the clinical investigation provisions of Directive 93/42/EEC (MDD) and introduced a more stringent, harmonised framework across all 27 EU member states. For sponsors running investigations in Spain, Germany, France, the Netherlands, or any other member state, understanding which pathway applies to their study is the single most consequential regulatory decision before CIP submission.
Complete guide to Phase I oncology trials in Western Europe in 2026: CTIS submissions, country selection (Spain, France, Germany, Netherlands), site criteria, study design, and CRO selection for biotech sponsors.
")
")